
Fabrication of Porous Carbon Films and Their Impact on Carbon/Polypropylene Interfacial Bonding


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Polymer Solution Preparation
2.3. Precursor Polymer Film Preparation
2.4. Carbon Film Fabrication
2.5. Materials Characterization
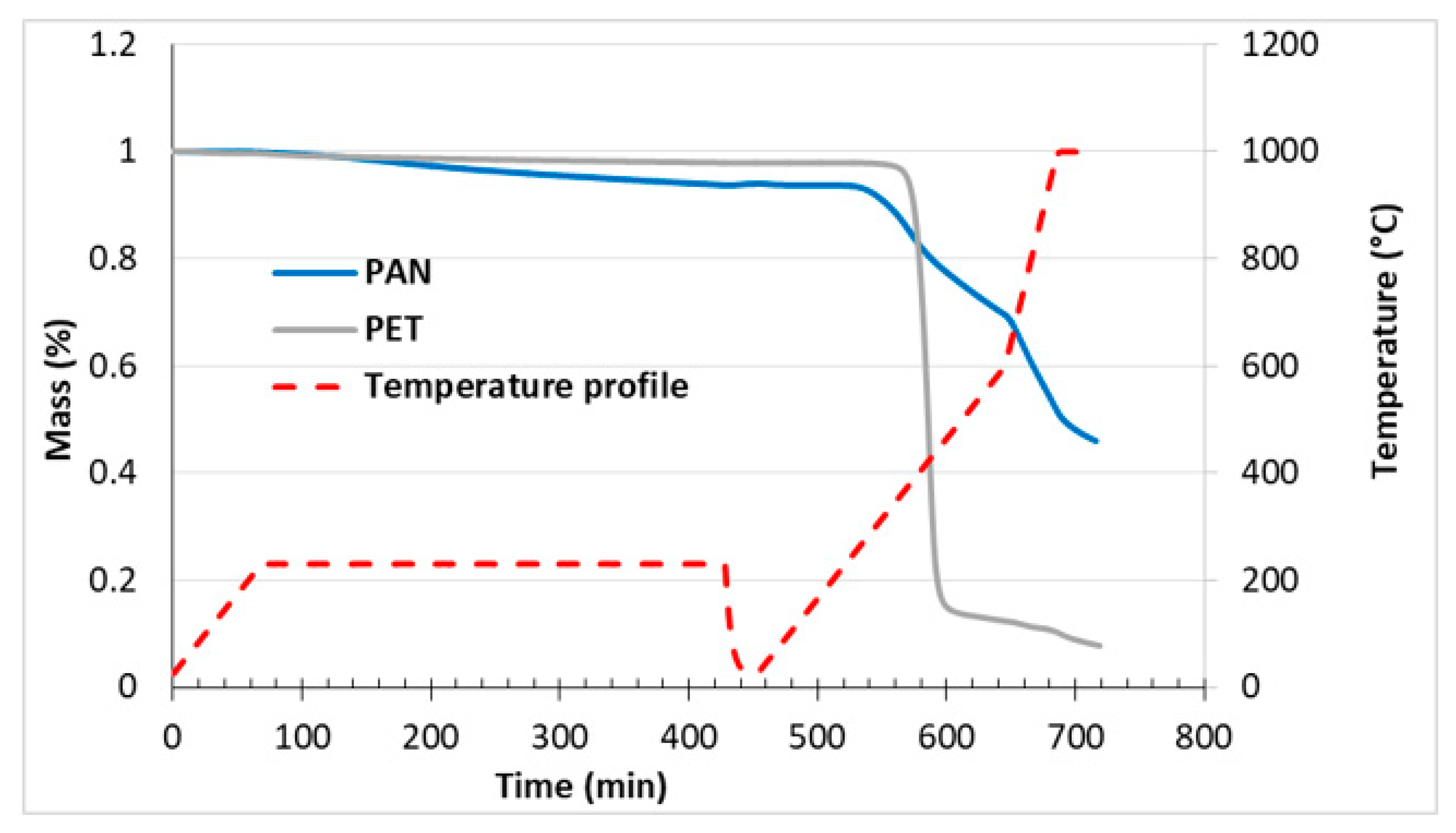
2.6. Thermogravimetric Analysis
2.7. Interlaminar Shear Strength Measurement
3. Result and Discussion
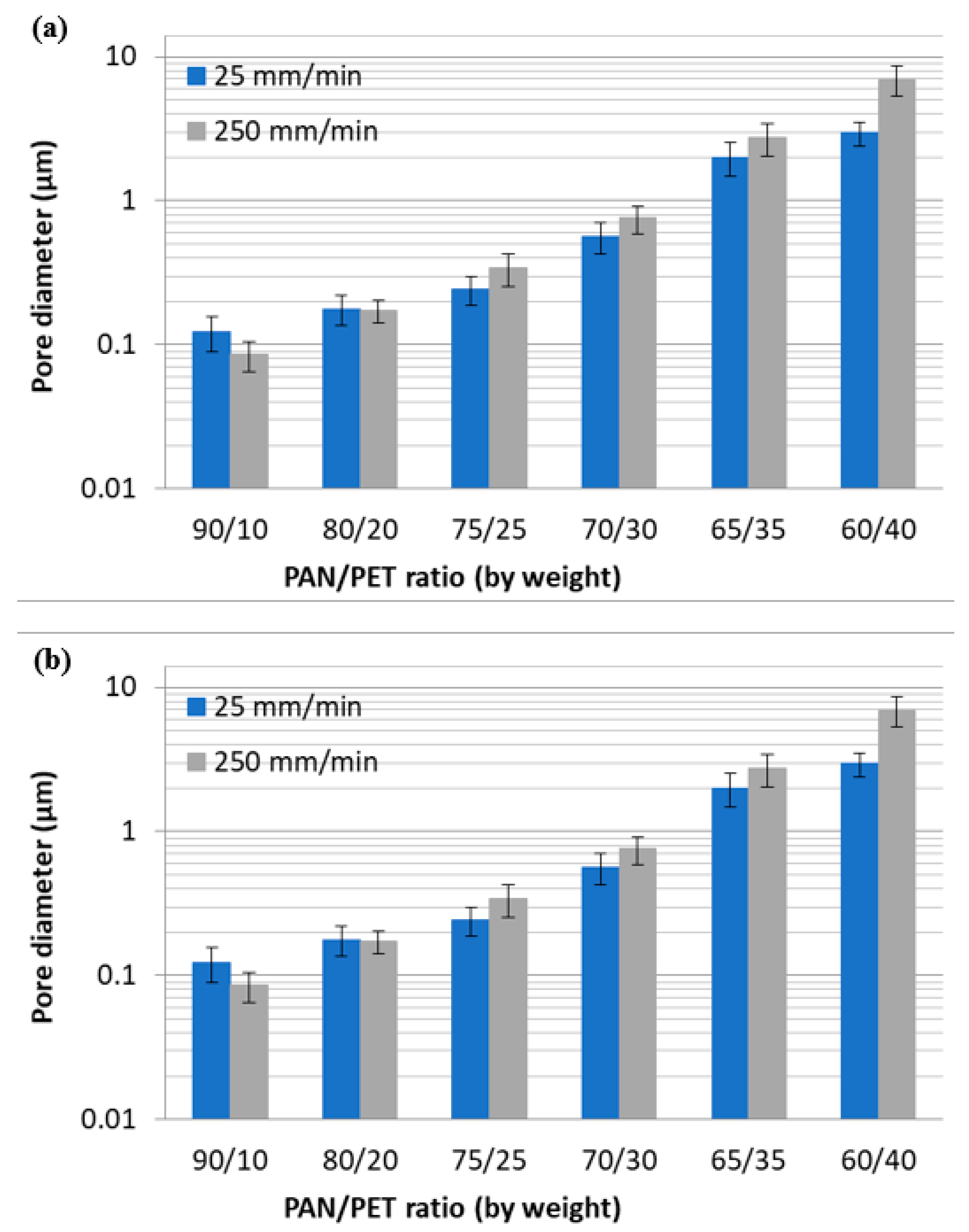
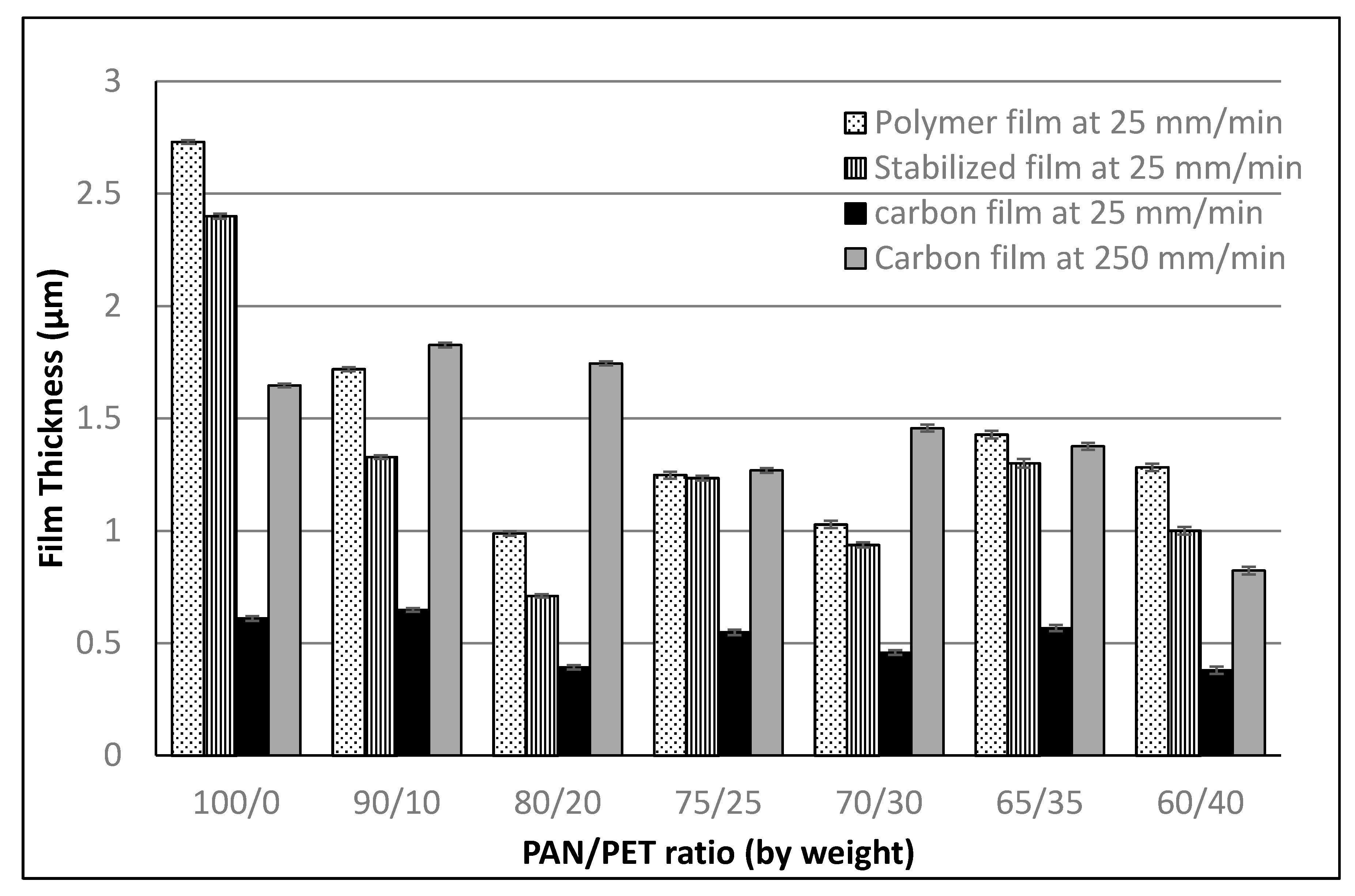
3.1. Fabrication and Morphology of Polymer Films
3.2. Fabrication and Morphology of Carbon Films
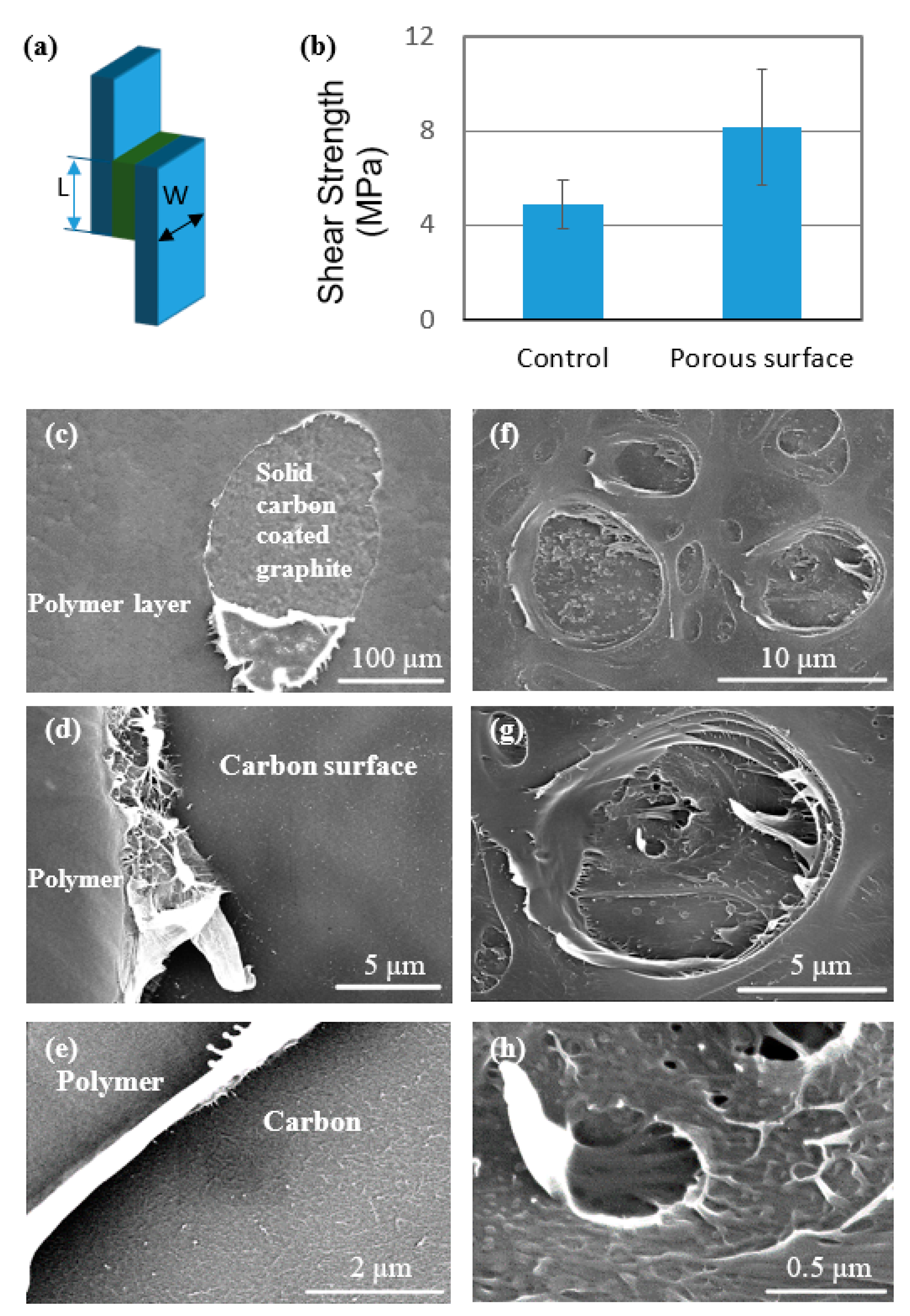
3.3. Interlaminar Shear Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bledzki, A.K.; Seidlitz, H.; Krenz, J.; Goracy, K.; Urbaniak, M.; Rosch, J.J. Recycling of Carbon Fiber Reinforced Composite Polymers-Review-Part 2: Recovery and Application of Recycled Carbon Fibers. Polymers 2020, 12, 3003. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Y.; Xu, L.; Liu, J.H.; Peng, J.H.; Han, Z.H.; Shen, Z.G.; Guo, S.H. Efficient Method of Recycling Carbon Fiber from the Waste of Carbon Fiber Reinforced Polymer Composites. Polym. Degrad. Stabil. 2020, 182. [Google Scholar] [CrossRef]
- Gopalraj, S.K.; Karki, T. A Review on the Recycling of Waste Carbon Fibre/Glass Fibre-Reinforced Composites: Fibre Recovery, Properties and Life-Cycle Analysis. Sn. Appl. Sci. 2020, 2, s42452-s020. [Google Scholar]
- Kiss, P.; Stadlbauer, W.; Burgstaller, C.; Stadler, H.; Fehringer, S.; Haeuserer, F.; Archodoulaki, V.M. In-House Recycling of Carbon- and Glass Fibre-Reinforced Thermoplastic Composite Laminate Waste into High-Performance Sheet Materials. Compos. Part A Appl. Sci. Manuf. 2020, 139. [Google Scholar] [CrossRef]
- Kumar, S.; Krishnan, S. Recycling of Carbon Fiber with Epoxy Composites by Chemical Recycling for Future Perspective: A Review. Chem. Pap. 2020, 74, 3785–3807. [Google Scholar] [CrossRef]
- Utekar, S.; Suriya, V.K.; More, N.; Rao, A. Comprehensive Study of Recycling of Thermosetting Polymer Composites - Driving Force, Challenges and Methods. Compos. Part B Eng. 2021, 207. [Google Scholar] [CrossRef]
- Zhang, J.; Chevali, V.S.; Wang, H.; Wang, C.H. Current Status of Carbon Fibre and Carbon Fibre Composites Recycling. Compos. Part. B Eng. 2020, 193. [Google Scholar] [CrossRef]
- Pimenta, S.; Pinho, S.T. Recycling Carbon Fibre Reinforced Polymers for Structural Applications: Technology Review and Market Outlook. Waste Manag. 2011, 31, 378–392. [Google Scholar] [CrossRef] [Green Version]
- Li, P.D.; Zhao, Y.; Long, X.; Zhou, Y.W.; Chen, Z.Y. Ductility Evaluation of Damaged Recycled Aggregate Concrete Columns Repaired With Carbon Fiber-Reinforced Polymer and Large Rupture Strain FRP. Front. Mater. 2020, 7. [Google Scholar] [CrossRef]
- Tapper, R.J.; Longana, M.L.; Norton, A.; Potter, K.D.; Hamerton, I. An Evaluation of Life Cycle Assessment and its Application to the Closed-Loop Recycling of Carbon Fibre Reinforced Polymers. Compos. Part. B Eng. 2020, 184. [Google Scholar] [CrossRef]
- Giorgini, L.; Benelli, T.; Brancolini, G.; Mazzocchetti, L. Recycling of Carbon Fiber Reinforced Composite Waste to Close Their Life Cycle in a Cradle-to-Cradle Approach. Curr. Opin. Green Sust. 2020, 26. [Google Scholar] [CrossRef]
- Liu, Z.; Turner, T.A.; Wong, K.H.; Pickering, S.J. Development of High Performance Recycled Carbon Fibre Composites with an Advanced Hydrodynamic Fibre Alignment Process. J. Clean Prod. 2021, 278. [Google Scholar] [CrossRef]
- Wellekotter, J.; Resch, J.; Baz, S.; Gresser, G.T.; Bonten, C. Insights into the Processing of Recycled Carbon Fibers via Injection Molding Compounding. J. Compos. Sci. 2020, 4, 161. [Google Scholar] [CrossRef]
- Ota, H.; Jespersen, K.M.; Saito, K.; Wada, K.; Okamoto, K.; Hosoi, A.; Kawada, H. Effect of the interfacial nanostructure on the Interlaminar Fracture Toughness and Damage Mechanisms of Directly Bonded Carbon Fiber Reinforced Thermoplastics and Aluminum. Compos. Part A Appl. Sci. Manuf. 2020, 139. [Google Scholar] [CrossRef]
- Khurshid, M.F.; Hengstermann, M.; Hasan, M.M.B.; Abdkader, A.; Cherif, C. Recent Developments in the Processing of Waste Carbon Fibre for Thermoplastic Composites—A Review. J. Compos. Mater. 2020, 54, 1925–1944. [Google Scholar] [CrossRef]
- Roux, M.; Eguemann, N.; Dransfeld, C.; Thiebaud, F.; Perreux, D. Thermoplastic Carbon Fibre-Reinforced Polymer Recycling with Electrodynamical Fragmentation: From Cradle to Cradle. J. Compos. 2017, 30, 381–403. [Google Scholar] [CrossRef]
- Hirayama, D.; Saron, C.; Botelho, E.C.; Costa, M.L.; Ancelotti, A.C. Polypropylene Composites Manufactured from Recycled Carbon Fibers from Aeronautic Materials Waste. Mater. Res.-Ibero-Am. J. 2017, 20, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, K. Carbon Fiber Reinforced Thermoplastic Composites for Future Automotive Applications. AIP Conf. Proc. 2016, 1736. [Google Scholar] [CrossRef]
- Stoeffler, K.; Andjelic, S.; Legros, N.; Roberge, J.; Schougaard, S.B. Polyphenylene Sulfide (PPS) Composites Reinforced with Recycled Carbon Fiber. Compos. Sci. Technol. 2013, 84, 65–71. [Google Scholar] [CrossRef]
- Sharma, M.; Gao, S.L.; Mader, E.; Sharma, H.; Wei, L.Y.; Bijwe, J. Carbon Fiber Surfaces and Composite Interphases. Compos. Sci. Technol. 2014, 102, 35–50. [Google Scholar] [CrossRef]
- Huang, X.S. Fabrication and Properties of Carbon Fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef]
- Donnet, J.-B.; Wang, T.K.; Peng, J.C.M.; Rebouillat, S. Carbon Fibers, 3rd ed.; Marcel Dekker Inc.: New York, NY, USA, 1998. [Google Scholar]
- Fu, S.Y.; Lauke, B.; Mäder, E.; Yue, C.Y.; Hu, X. Tensile Properties of Short-Glass-Fiber- and Short-Carbon-Fiber-Reinforced Polypropylene Composites. Compos. Part A Appl. Sci. Manuf. 2000, 31, 1117–1125. [Google Scholar] [CrossRef]
- Gamze Karsli, N.; Aytac, A.; Akbulut, M.; Deniz, V.; Güven, O. Effects of Irradiated Polypropylene Compatibilizer on the Properties of Short Carbon Fiber Reinforced Polypropylene Composites. Radiat. Phys. Chem. 2013, 84, 74–78. [Google Scholar] [CrossRef]
- Han, S.H.; Oh, H.J.; Kim, S.S. Evaluation of Fiber Surface Treatment on the Interfacial Behavior of Carbon Fiber-Reinforced Polypropylene Composites. Compos. Part. B Eng. 2014, 60, 98–105. [Google Scholar] [CrossRef]
- Karsli, N.G.; Aytac, A. Effects of Maleated Polypropylene on the Morphology, Thermal and Mechanical Properties of Short Carbon Fiber Reinforced Polypropylene Composites. Mater. Des. 2011, 32, 4069–4073. [Google Scholar] [CrossRef]
- Rezaei, F.; Yunus, R.; Ibrahim, N.A. Effect of Fiber Length on Thermomechanical Properties of Short Carbon Fiber Reinforced Polypropylene Composites. Mater. Des. 2009, 30, 260–263. [Google Scholar] [CrossRef]
- Tian, H.; Yao, Y.; Liu, D.; Li, Y.; Jv, R.; Xiang, G.; Xiang, A. Enhanced Interfacial Adhesion and Properties of Polypropylene/Carbon Fiber Composites by Fiber Surface Oxidation in Presence of a Compatibilizer. Polym. Compos. 2019, 40, E654–E662. [Google Scholar] [CrossRef]
- Tian, H.F.; Yao, Y.Y.; Wang, C.Y.; Jv, R.; Ge, X.; Xiang, A.M. Essential Work of Fracture Analysis for Surface Modified Carbon Fiber/Polypropylene Composites with Different Interfacial Adhesion. Polym. Compos. 2020, 41, 3541. [Google Scholar] [CrossRef]
- Unterweger, C.; Duchoslav, J.; Stifter, D.; Fürst, C. Characterization of Carbon Fiber Surfaces and Their Impact on the Mechanical Properties of Short Carbon Fiber Reinforced Polypropylene Composites. Compos. Sci. Technol. 2015, 108, 41–47. [Google Scholar] [CrossRef]
- Vishkaei, M.S.; Salleh, M.A.M.; Yunus, R.; Biak, D.R.A.; Danafar, F.; Mirjalili, F. Effect of Short Carbon Fiber Surface Treatment on Composite Properties. J. Compos. Mater. 2010, 45, 1885–1891. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; He, X.; Nie, M.; Wang, Q. Mechanical Interlock Effect Between Polypropylene/Carbon Fiber Composite Generated by Interfacial Branched Fibers. Compos. Sci. Technol. 2018, 167, 1–6. [Google Scholar] [CrossRef]
- Huang, Y.L.; Young, R.J. Interfacial Micromechanics in Thermoplastic and Thermosetting Matrix Carbon Fibre Composites. Compos. Part A Appl. Sci. Manuf. 1996, 27, 973–980. [Google Scholar] [CrossRef]
- Tang, L.G.; Kardos, J.L. A Review of Methods for Improving the Interfacial Adhesion between Carbon Fiber and Polymer Matrix. Polym. Compos. 1997, 18, 100–113. [Google Scholar] [CrossRef]
- Gravis, D.; Moisan, S.; Poncin-Epaillard, F. Characterization of surface physico-chemistry and morphology of plasma-sized carbon fiber. Thin Solid Films 2021, 721, 11. [Google Scholar] [CrossRef]
- Donnet, J.B.; Brendle, M.; Dhami, T.L.; Bahl, O.P. Plasma Treatment Effect on the Surface-Energy of Carbon and Carbon-Fibers. Carbon 1986, 24, 757–770. [Google Scholar] [CrossRef]
- Donnet, J.B.; Cazeneuve, C.; Schultz, J.; Shanahan, M.E.R. Surface-Energy of Carbon-Fibers. Carbon 1980, 18, 61. [Google Scholar]
- Donnet, J.B.; Dhami, T.L.; Dong, S.; Brendle, M. Microwave Plasma Treatment Effect on the Surface-Energy of Carbon-Fibers. J. Phys. D Appl. Phys. 1987, 20, 269–275. [Google Scholar] [CrossRef]
- Donnet, J.B.; Ehrburger, P. Carbon-Fiber in Polymer Reinforcement. Carbon 1977, 15, 143–152. [Google Scholar] [CrossRef]
- Donnet, J.B.; Guilpain, G. Surface Treatments and Properties of Carbon-Fibers. Carbon 1989, 27, 749–757. [Google Scholar] [CrossRef]
- Donnet, J.B.; Guilpain, G. Surface Characterization of Carbon-Fibers. Composites 1991, 22, 59–62. [Google Scholar] [CrossRef]
- Ehrburger, P.; Herque, J.J.; Donnet, J.P. Recent Developments in Carbon-Fiber Treatments. Acs. Sym. Ser. 1976, 324–334. [Google Scholar]
- Sadeghvishakei, M.; Yunus, R.; Salleh, M.A.M.; Pignolet, A. Mechanical Investigation in Whiskerized Carbon Fiber/Polypropylene Composite. In Proceedings of the Smart Materials, Structures & NDTin Aerospace, Montreal, QC Canada, November 2011. [Google Scholar]
- Zhang, Q.H.; Liu, J.W.; Sager, R.; Dai, L.M.; Baur, J. Hierarchical Composites of Carbon Nanotubes on Carbon Fiber: Influence of Growth Condition on Fiber Tensile Properties. Compos. Sci. Technol. 2009, 69, 594–601. [Google Scholar] [CrossRef]
- Peng, Y.C.; Burtovyy, R.; Yang, Y.; Urban, M.W.; Kennedy, M.S.; Kornev, K.G.; Bordia, R.; Luzinov, I. Towards scalable Fabrication of Ultrasmooth and Porous thin Carbon Films. Carbon 2016, 96, 184–195. [Google Scholar] [CrossRef]
- Fried, J. Polymer Science and Technology, 3rd ed.; Pearson: London, UK, 2014. [Google Scholar]
- Seeber, M.; Zdyrko, B.; Burtovvy, R.; Andrukh, T.; Tsai, C.-C.; Owens, J.R.; Kornev, K.G.; Luzinov, I. Surface Grafting of Thermoresponsive Microgel Nanoparticles. Soft Matter. 2011, 7, 9962–9971. [Google Scholar] [CrossRef]
- Walheim, S.; Böltau, M.; Mlynek, J.; Krausch, G.; Steiner, U. Structure Formation via Polymer Demixing in Spin-Cast Films. Macromolecules 1997, 30, 4995–5003. [Google Scholar] [CrossRef]
- Elbs, H.; Fukunaga, K.; Stadler, R.; Sauer, G.; Magerle, R.; Krausch, G. Microdomain Morphology of Thin ABC Triblock Copolymer Films. Macromolecules 1999, 32, 1204–1211. [Google Scholar] [CrossRef]
- Landau, L.; Levich, B. Dragging of a Liquid by a Moving Plate. Acta. Phys. URSS 1942, 17, 42–54. [Google Scholar]
- Mayer, H.C.; Krechetnikov, R. Landau-Levich Flow Visualization: Revealing the Flow Topology Responsible for the Film Thickening Phenomena. Phys. Fluids 2012, 24. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Polymer Concentration in Solution (wt.%) | PAN/PET (by Weight) | Dip-Coating Withdrawal Speed (mm/min) |
|---|---|---|---|
| PAN-3-25 | 3 | 100/0 | 25 |
| PAN-3-250 | 3 | 100/0 | 250 |
| PAN/PET-10-25 | 3 | 90/10 | 25 |
| PAN/PET-20-25 | 3 | 80/20 | 25 |
| PAN/PET-25-25 | 3 | 75/25 | 25 |
| PAN/PET-30-25 | 3 | 70/30 | 25 |
| PAN/PET-35-25 | 3 | 65/35 | 25 |
| PAN/PET-40-25 | 3 | 60/40 | 25 |
| PAN/PET-10-250 | 3 | 90/10 | 250 |
| PAN/PET-20-250 | 3 | 80/20 | 250 |
| PAN/PET-25-250 | 3 | 75/25 | 250 |
| PAN/PET-30-250 | 3 | 70/30 | 250 |
| PAN/PET-35-250 | 3 | 65/35 | 250 |
| PAN/PET-40-250 | 3 | 60/40 | 250 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Burtovyy, R.; Bordia, R.; Luzinov, I. Fabrication of Porous Carbon Films and Their Impact on Carbon/Polypropylene Interfacial Bonding. J. Compos. Sci. 2021, 5, 108. https://doi.org/10.3390/jcs5040108
Peng Y, Burtovyy R, Bordia R, Luzinov I. Fabrication of Porous Carbon Films and Their Impact on Carbon/Polypropylene Interfacial Bonding. Journal of Composites Science. 2021; 5(4):108. https://doi.org/10.3390/jcs5040108
Chicago/Turabian StylePeng, Yucheng, Ruslan Burtovyy, Rajendra Bordia, and Igor Luzinov. 2021. "Fabrication of Porous Carbon Films and Their Impact on Carbon/Polypropylene Interfacial Bonding" Journal of Composites Science 5, no. 4: 108. https://doi.org/10.3390/jcs5040108
APA StylePeng, Y., Burtovyy, R., Bordia, R., & Luzinov, I. (2021). Fabrication of Porous Carbon Films and Their Impact on Carbon/Polypropylene Interfacial Bonding. Journal of Composites Science, 5(4), 108. https://doi.org/10.3390/jcs5040108