
First-Principles Calculations of Crystallographic and Electronic Structural Properties of Au-Cu Alloys




Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
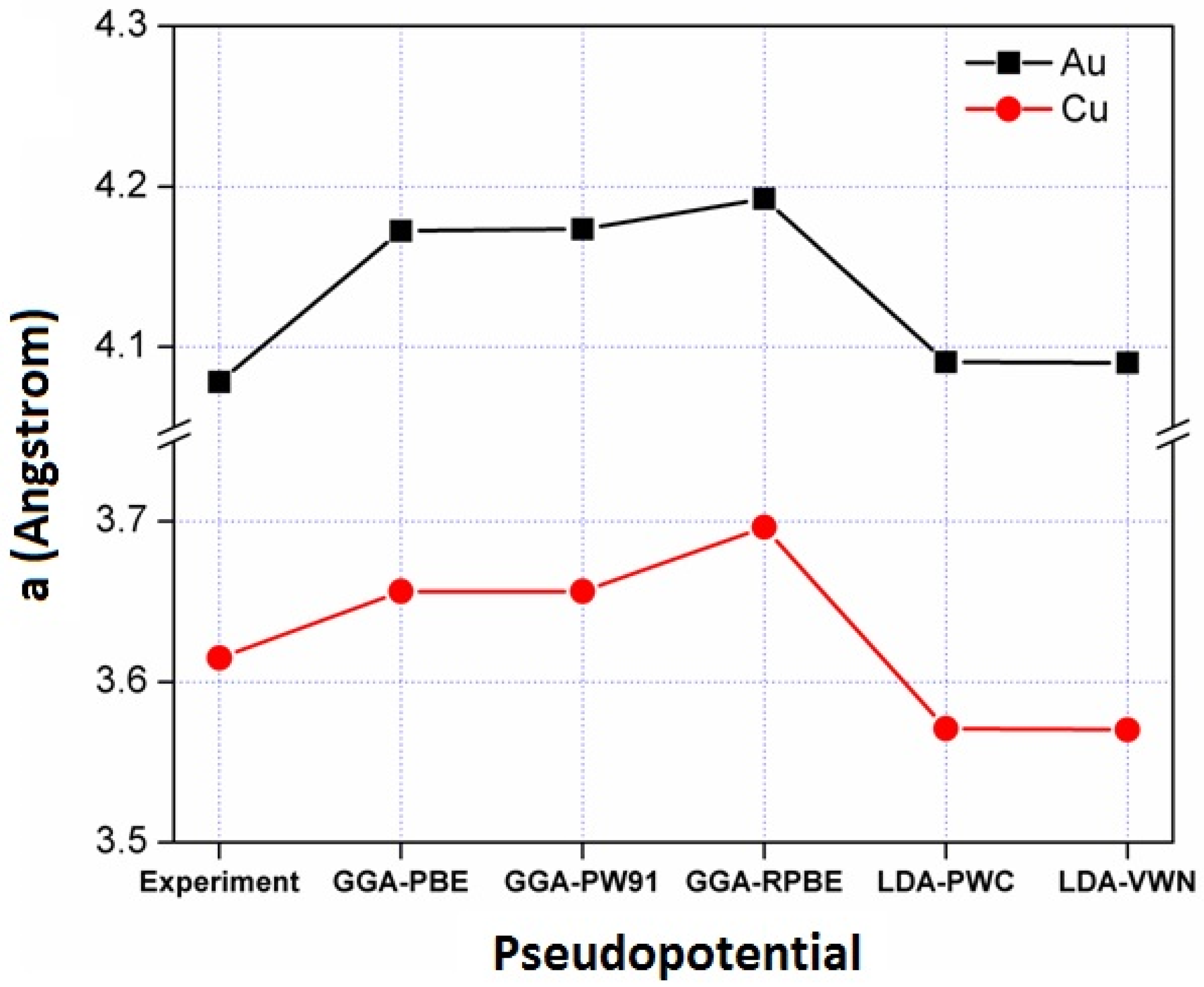
3.1. Pseudo-Potential Selection
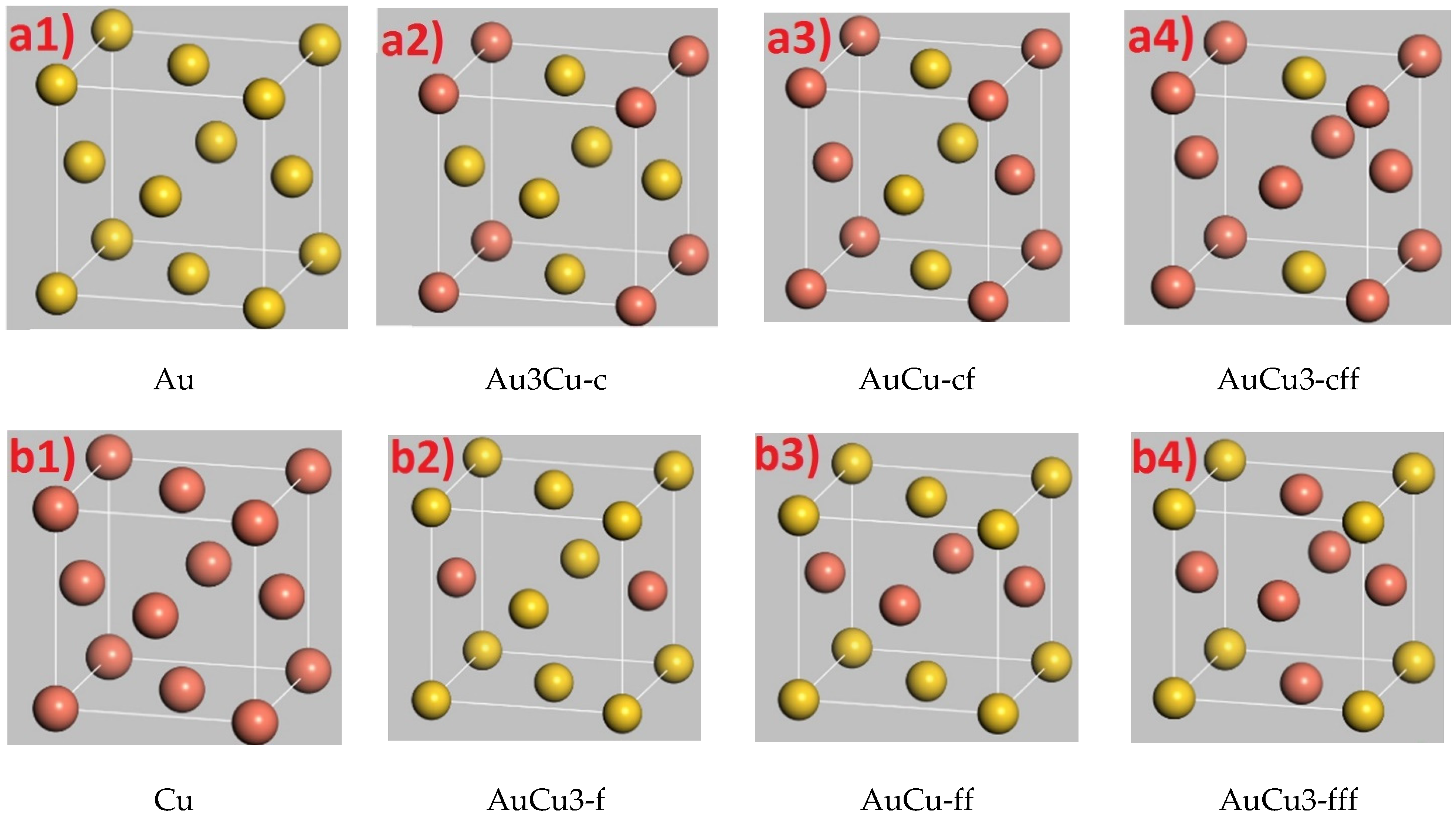
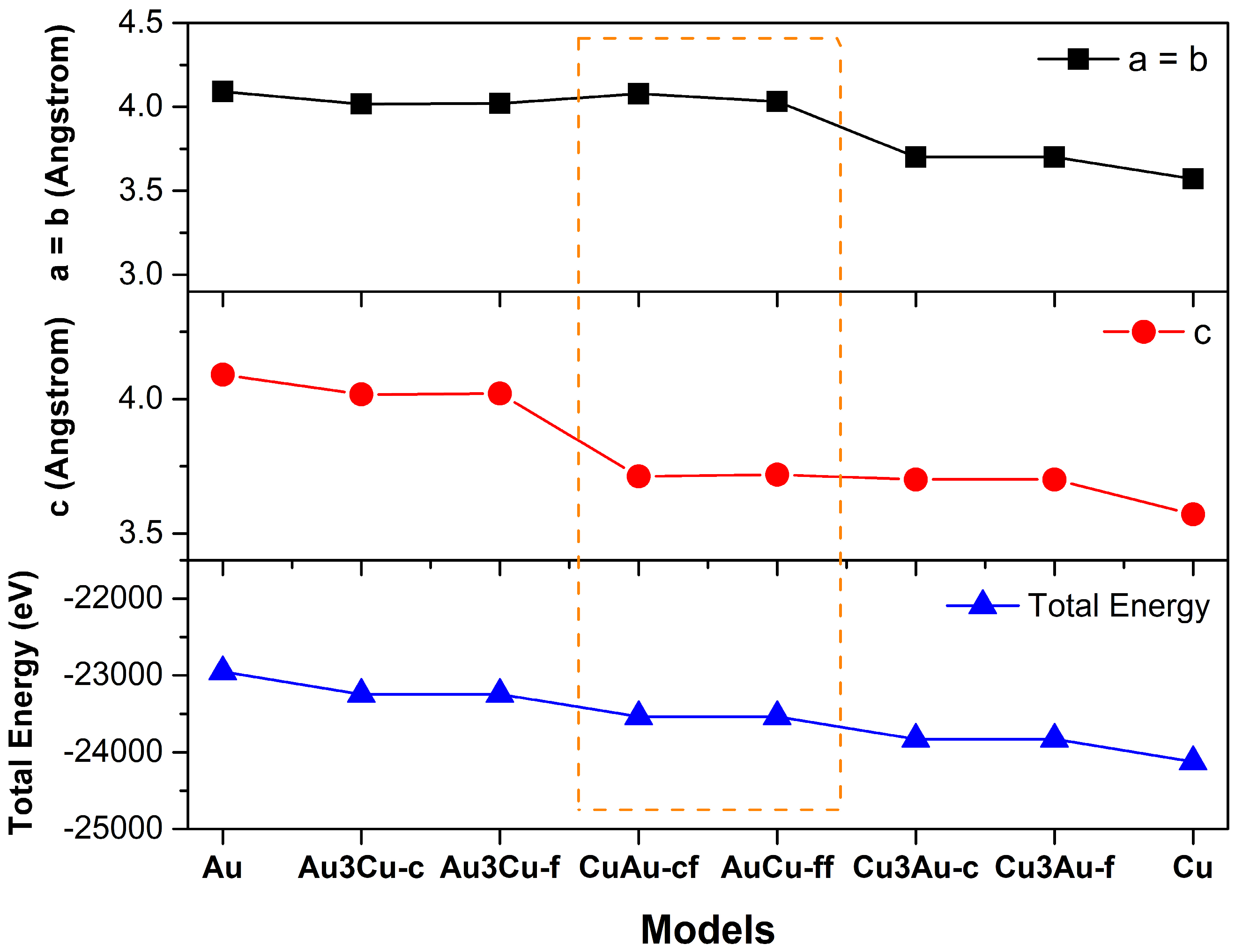
3.2. Formation of Au-Cu Alloys
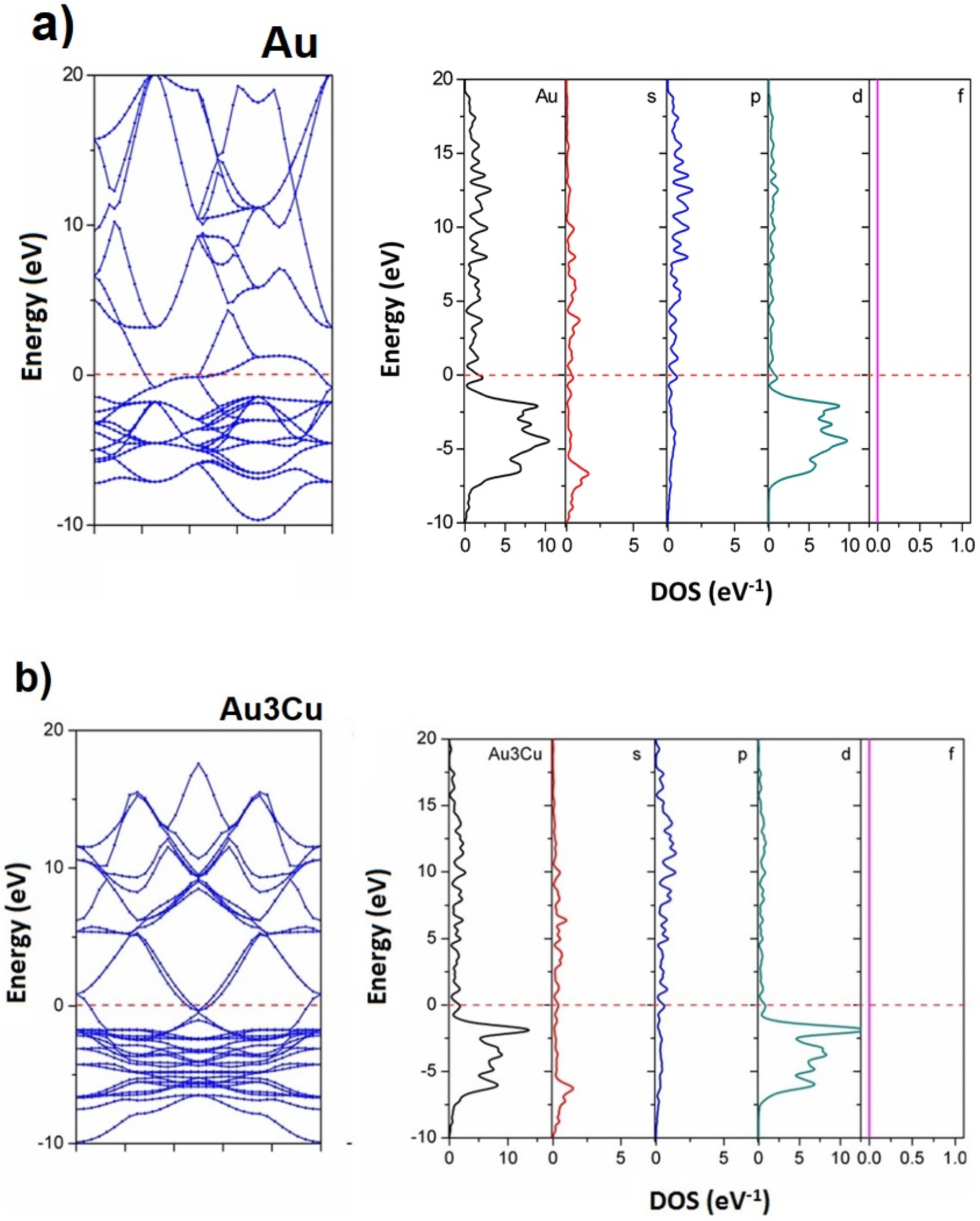
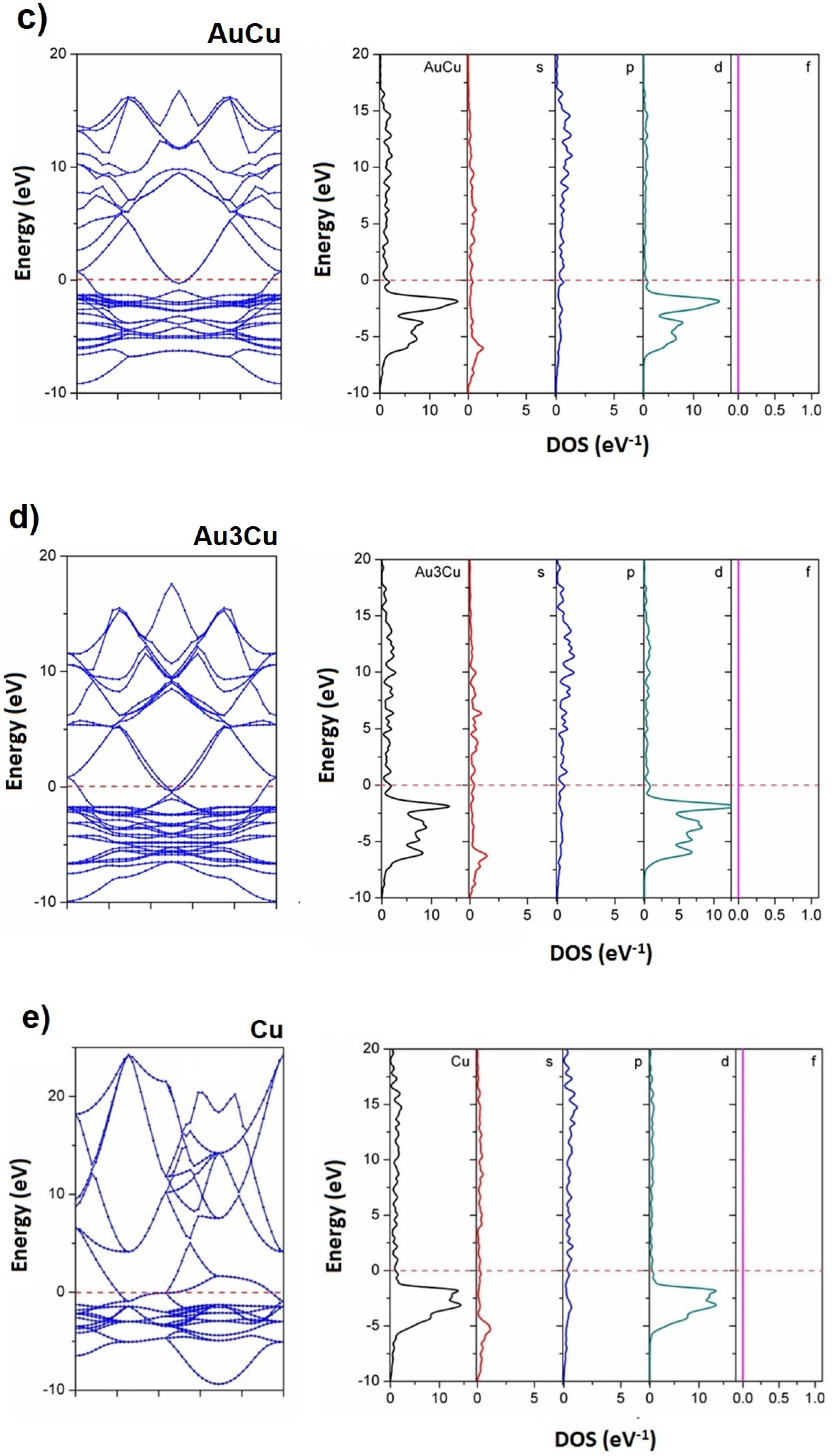
3.3. Electronic Structures of Au, Cu, and Au-Cu Alloys
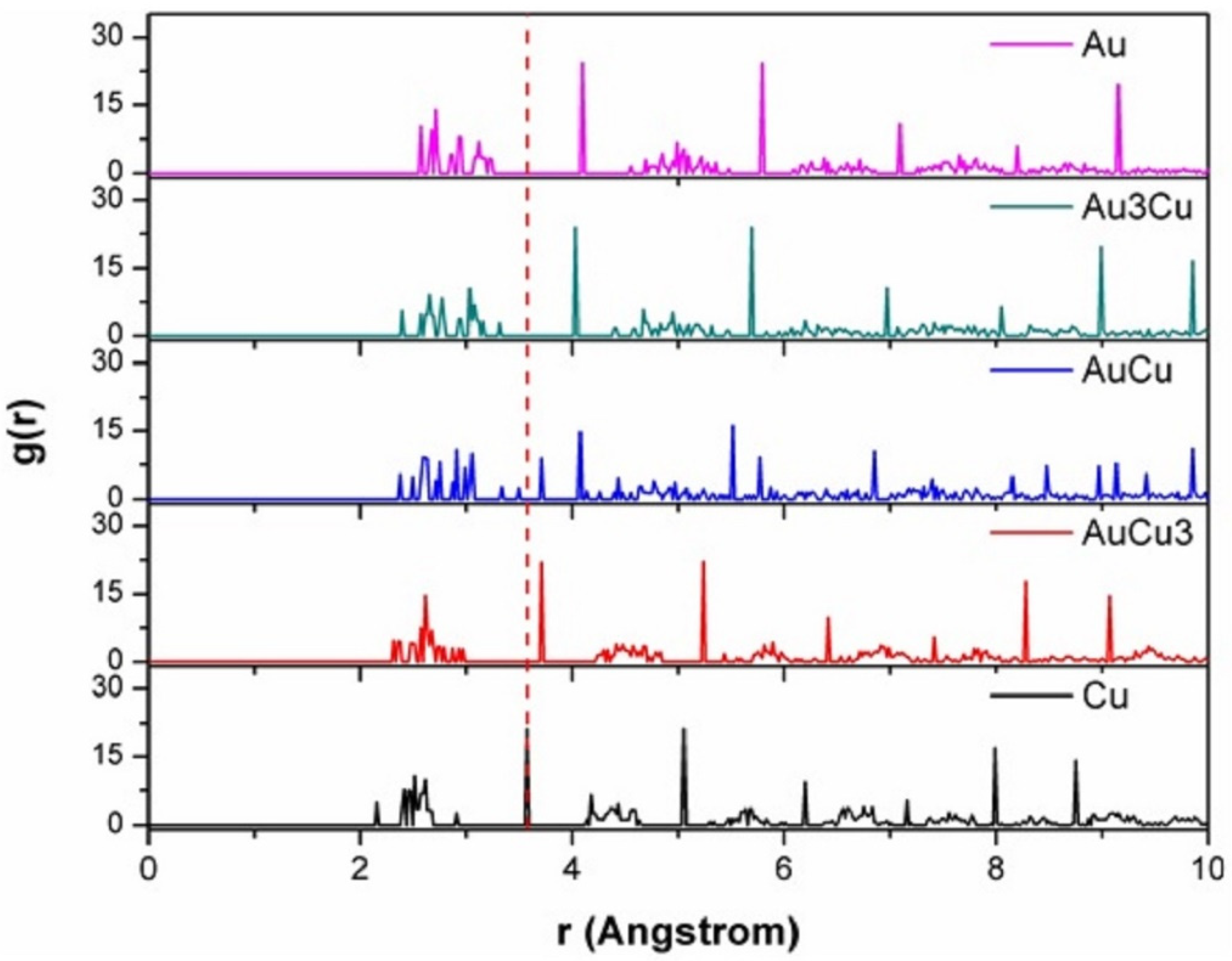
3.4. RDF Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tuan, T.Q.; Dung, N.T. Effect of heating rate, impurity concentration of Cu, atomic number, temperatures, time annealing temperature on the structure, crystallization temperature and crystallization process of Ni1−xCux bulk; x = 0.1, 0.3, 0.5, 0.7. Int. J. Mod. Phys. B 2018, 32, 1830009. [Google Scholar] [CrossRef]
- Dung, N.T.; Phuong, N.T. Molecular dynamic study on factors influencing the structure, phase transition and crystallization process of NiCu6912 nanoparticle. Mater. Chem. Phys. 2020, 250, 123075. [Google Scholar]
- Dung, N.T.; Van, C.L. Factors affecting the depth of the Earth’s surface on the heterogeneous dynamics of Cu1-xNix alloy, x =0.1, 0.3, 0.5, 0.7, 0.9 by Molecular Dynamics simulation method. Mater. Today Commun. 2021, 29, 102812. [Google Scholar]
- Hoc, N.Q.; Viet, L.H.; Dung, N.T. On the Melting of Defective FCC Interstitial Alloy c–FeC under Pressure up to 100 GPa. J. Electron. Mater. 2020, 49, 910–916. [Google Scholar] [CrossRef]
- Trung, V.Q.; Hung, H.M.; Khoe, L.V.; Duc, L.M.; Viet, N.T.B.; Linh, D.K.; Huong, V.T.; Dat, N.D.; Oanh, D.T.Y.; Luong, N.X.; et al. Synthesis and Characterization of Polypyrrole Film Doped with Both Molybdate and Salicylate and Its Application in the Corrosion Protection for Low Carbon Steel. ACS Omega 2022, 7, 19842–19852. [Google Scholar] [CrossRef]
- Luo, Y.; Qiu, Z.; Song, S.; Dong, S.; Wang, H.; Yu, H. Study on the sensing capability of Fe-C alloycorrosion sensing film. Mater. Corros. 2007, 58, 198–201. [Google Scholar] [CrossRef]
- Dung, N.T. Influence of impurity concentration, atomic number, temperature and tempering time on microstructure and phase transformation of Ni1−xFex (x = 0.1, 0.3, 0.5) nanoparticles. Mod. Phys. Lett. B 2018, 32, 1850204. [Google Scholar] [CrossRef]
- Dung, N.T.; Kien, P.H.; Phuong, N.T. Simulation on the Factors Affecting the Crystallization Process of FeNi Alloy by Molecular Dynamics. ACS Omega 2019, 4, 14605–14612. [Google Scholar]
- Dung, N.T.; Phuong, N.T. Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles. J. Alloys Compd. 2020, 812, 1521332. [Google Scholar]
- Dung, N.T.; Van, C.L.; Ţălu, Ş. The Structure and Crystallizing Process of NiAu Alloy: A Molecular Dynamics Simulation Method. J. Compos. Sci. 2021, 5, 18. [Google Scholar]
- Quoc, T.T.; Long, V.C.; Ţălu, Ş.; Dung, N.T. Molecular Dynamics Study on the Crystallization Process of Cubic Cu–Au Alloy. Appl. Sci. 2022, 12, 946. [Google Scholar] [CrossRef]
- Dung, N.T.; Cuong, N.C.; Van, D.Q. Study on the effect of doping on lattice constant and electronic structure of bulk AuCu by the density functional theory. J. Multiscale Model. 2020, 11, 2030001. [Google Scholar]
- Hoc, N.Q.; Hien, N.D.; Dung, N.T.; Van, C.L.; Ţălu, Ş. Study on the Melting Temperature, the Jumps of Volume, Enthalpy and Entropy at Melting Point, and the Debye Temperature for the BCC Defective and Perfect Interstitial Alloy WSi under Pressure. J. Compos. Sci. 2021, 5, 153. [Google Scholar]
- Van, C.L.; Umut, S.; Baykul, M.C.; Luong, D.T.; Țălu, Ș.; Dung, N.T. Electrochemical Deposition of Fe–Co–Ni Samples with Different Co Contents and Characterization of Their Microstructural and Magnetic Properties. Coatings 2022, 12, 346. [Google Scholar]
- Dung, N.T.; Phuong, N.T. Understanding the heterogeneous kinetics of Al nanoparticles by simulations method. J. Mol. Struct. 2020, 1218, 1284982. [Google Scholar]
- Dung, N.T. Z-AXIS deformation method to investigate the influence of system size, structure phase transition on mechanical properties of bulk nickel. Mater. Chem. Phys. 2020, 252, 123275. [Google Scholar]
- Liang, R.; Tian, R.; Ma, L.; Zhang, L.; Hu, Y.; Wang, J.; Wei, M.; Yan, D.; Evans, D.G.; Duan, X. A Supermolecular Photosensitizer with Excellent Anticancer Performance in Photodynamic Therapy. Adv. Funct. Mater. 2014, 24, 3144. [Google Scholar] [CrossRef]
- Shan, R.R.; Yan, L.G.; Yang, K.; Hao, Y.F.; Du, B. Adsorption of Cd(II) by Mg−Al−CO3 and magnetic Fe3O4/Mg−Al−CO3 layered double hydroxides: Kinetic, isothermal, thermodynamic and mechanistic studies. J. Hazard. Mater. 2015, 299, 42–49. [Google Scholar] [CrossRef]
- Luo, J.; Jeong-Hyeok, I.; Mayer, M.T.; Schreier, M.; Nazeeruddin, M.K.; David Tilley, S.; Fan, H.J.; Gratzel, M. Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earthabundant catalysts. Science 2014, 345, 1593–1596. [Google Scholar] [CrossRef]
- Ross, C. Patterned magnetic recording media. Annu. Rev. Mater. Res. 2001, 31, 203. [Google Scholar] [CrossRef]
- Lin, F.H.; Doong, R.A. Bifunctional Au-Fe3O4 Heterostructures for Magnetically Recyclable Catalysis of Nitroph enol Reduction. J. Phys. Chem. C 2011, 115, 6591. [Google Scholar] [CrossRef]
- Kim, Y.; Johnson, R.C.; Hupp, J.T. Gold Nanoparticle-Based Sensing of “Spectroscopically Silent” Heavy Metal Ions. Nano Lett. 2001, 1, 165–167. [Google Scholar] [CrossRef]
- Son, S.J.; Reichel, J.; He, B.; Schuchman, M.; Lee, S.B. Magnetic Nanotubes for Magnetic-Field-Assisted Bioseparation, Biointeraction, and Drug Delivery. J. Am. Chem. Soc. 2005, 127, 7316–7317. [Google Scholar] [CrossRef] [PubMed]
- Aish, M.M. Mechanical properties and sound velocity of gold copper (AuCu) II superlattice: 3D molecular dynamic (MD) simulation. J. Theor. Appl. Mech. 2020, 58, 901–909. [Google Scholar] [CrossRef]
- Han, X.J.; Chen, M.; Guo, Z.Y. Thermophysical properties of undercooled liquid Au–Cu alloys from molecular dynamics simulations. J. Phys. Condens. Matter. 2004, 16, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Kart, H.H.; Tomak, M.; Çağın, T. Thermal and mechanical properties of Cu–Au intermetallic alloys. Model. Simul. Mater. Sci. Eng. 2005, 13, 657–669. [Google Scholar] [CrossRef]
- Barrera, G.D.; de Tendler, R.H.; Isoardi, E.P. Structure and energetics of Cu-Au alloys. Model. Simul. Mater. Sci. Eng. 2000, 8, 389. [Google Scholar] [CrossRef]
- Wang, J.; Qin, H.; Chen, J.; Yang, D.; Zhang, G. First-Principles Study on the Elastic Mechanical Properties and Anisotropies of Gold–Copper Intermetallic Compounds. Metals 2022, 12, 959. [Google Scholar] [CrossRef]
- Maier, S.A.; Brongersma, M.L.; Kik, P.G.; Meltzer, S.; Requicha, A.A.; Atwater, H.A. Plasmonics—A route to nanoscale optical devices. Adv. Mater. 2001, 13, 1501–1505. [Google Scholar] [CrossRef]
- Alivov, Y.; Fan, Z.Y. Efficiency of dye sensitized solar cells based on TiO2 nanotubes filled with nanoparticles. Appl. Phys. Lett. 2009, 95, 063504. [Google Scholar] [CrossRef]
- Kneipp, K.; Wang, Y.; Kneipp, H.; Perelman, L.T.; Itzkan, I.; Dasari, R.R.; Feld, M.S. Single molecule detection using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Lal, S.; Grady, N.K.; Kundu, J.; Levin, C.S.; Lassiter, J.B.; Halas, N.J. Tailoring plasmonic substrates for surface enhanced spectroscopies. Chem. Soc. Rev. 2008, 37, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Muňoz, H.R.; Mejίa-Rosales, S. The AuCu Phase Diagram at the Nano Scale: A Molecular Dynamics Approach. J. Clust. Sci. 2022, 33, 785–793. [Google Scholar] [CrossRef]
- Kong, G.X.; Ma, X.J.; Liu, Q.J.; Li, Y.; Liu, Z.T. Structural stability, elastic and thermodynamic properties of Au–Cu alloys from first-principles calculations. Physica B Condens. Matter. 2018, 533, 58–62. [Google Scholar] [CrossRef]
- Aslanides, A.; Hayoun, M.; Pontikis, V. Atomic structure of low-index and (11n) surfaces in ordered Cu3Au. Surf. Sci. 1997, 370, L163–L167. [Google Scholar] [CrossRef]
- Țălu, Ș.; Yadav, R.P.; Mittal, A.K.; Arman, A.; Luna, C.; Mardani, M.; Solaymani, S.; Arman, A.; Hafezi, F.; Ahmadpourian, A.; et al. Application of Mie theory and fractal models to determine the optical and surface roughness of Ag-Cu thin films. Opt. Quantum Electron. 2017, 49, 256. [Google Scholar] [CrossRef]
- Țălu, Ș. Micro and Nanoscale Characterization of Threedimensional Surfaces: Basics and Applications; Napoca Star Publishing House: Cluj-Napoca, Romania, 2015. [Google Scholar]
- Zeng, D.; Yang, L.; Zhou, P.; Hu, D.; Xie, Y.; Li, S.; Jiang, L.; Ling, Y.; Zhao, J. A-Cu alloys deposited on titanium dioxide nanosheets for efficient photocatalytic hydrogen evolution. Int. J. Hydrogen Energy 2018, 43, 15155–15163. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V.; Zunger, A. First-principles theory of short-range order in size-mismatched metal alloys: Cu-Au, Cu-Ag, and Ni-Au. Phys. Rev. B 1998, 57, 4332–4348. [Google Scholar] [CrossRef]
- Zhang, B.; Edmund, T.; Shu, X.; Hu, W.; Deng, H. Simulation calculations of surface segregation for Au–Cu alloys using an analytic embedded atom model. Phys. Status Solidi (A) 2005, 202, 2686–2699. [Google Scholar]
- Okada, M.; Tsuda, Y.; Oka, K.; Kojima, K.; Diño, W.A.; Yoshigoe, A.; Kasai, H. Experimental and Theoretical Studies on Oxidation of Cu-Au Alloy Surfaces: Effect of Bulk Au Concentration. Sci. Rep. 2016, 6, 31101. [Google Scholar] [CrossRef] [Green Version]
- Jahelnabi, M.E.; Eisa, M.H.; Alobaid, R.A.; Bush, H.S. First-Principles Molecular Mechanics Calculations of the Lattice Constant and Bulk Modulus for Au-Cu Nano-Alloy. Int. J. Sci. Res. (IJSR) 2018, 7, 94–97. [Google Scholar]
- Levämäki, H.; Tian, L.; Kokko, K.; Vitos, L. Gradient-level and nonlocal density functional descriptions of Cu-Au intermetallic compounds. Eur. Phys. J. B 2018, 91, 128. [Google Scholar] [CrossRef]
- Tian, L.Y.; Levämäki, H.; Kuisma, M.; Kokko, K.; Nagy, Á.; Vitos, L. Density functional theory description of random Cu-Au alloys. Phys. Rev. B 2019, 99, 064202. [Google Scholar] [CrossRef] [Green Version]
- von Boehm, J.; Nieminen, R.M. Molecular-dynamics study of partial edge dislocations in copper and gold: Interactions, structures, and self-diffusion. Phys. Rev. B 1996, 53, 8956–8966. [Google Scholar] [CrossRef] [Green Version]
- Van, C.L.; Van, D.Q.; Dung, N.T. Ab Initio Calculations on the Structural and Electronic Properties of AgAu Alloys. ACS Omega 2020, 5, 31391–31397. [Google Scholar]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Hamann, D.R.; Schlüter, M.; Chiang, C. Norm-Conserving Pseudopotentials. Phys. Rev. Lett. 1979, 43, 1494–1497. [Google Scholar] [CrossRef]
- Straumanis, M.E.; Yu, L.S. Lattice parameters, densities, expansion coefficients and perfection of structure of Cu and of Cu-In [alpha] phase. Acta Cryst. A 1969, 25, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.C.; Li, S.H.; Lin, S.W. Effect of Fe Concentration on Fe-Doped Anatase TiO2 from GGA+U Calculations. Int. J. Photoenergy 2012, 2012, 823498. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Au | Au3Cu | AuCu | AuCu3 | Cu | |
|---|---|---|---|---|---|
| Bravais Lattice | FCC | Triclinic | Triclinic | Triclinic | FCC |
| a = b (Å) | 4.0910 | 4.0170 | 3.7185 | 3.7002 | 3.5710 |
| c (Å) | 4.0910 | 4.0170 | 4.0325 | 3.7002 | 3.5710 |
| Total Energy Etot (eV) | −22,952 | −23,245 | −23,538 | −23,830 | −24,124 |
| Models | Au | Au3Cu | AuCu | AuCu3 | Cu |
|---|---|---|---|---|---|
| m | 0 | 1 | 2 | 3 | 4 |
| n | 0 | 1 | 2 | 3 | 4 |
| Formation Energy (×10−2 eV) | 0 | 0.544 | 11.973 | 97.143 | 0 |
| Models | VB Widths | CB Widths |
|---|---|---|
| Au | 10.3945 | 23.2789 |
| Au3Cu | 10.3154 | 20.1977 |
| AuCu | 10.2024 | 21.1547 |
| Cu3Au | 11.0008 | 22.9690 |
| Cu | 10.3949 | 23.2789 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trong, D.N.; Long, V.C.; Saraç, U.; Quoc, V.D.; Ţălu, Ş. First-Principles Calculations of Crystallographic and Electronic Structural Properties of Au-Cu Alloys. J. Compos. Sci. 2022, 6, 383. https://doi.org/10.3390/jcs6120383
Trong DN, Long VC, Saraç U, Quoc VD, Ţălu Ş. First-Principles Calculations of Crystallographic and Electronic Structural Properties of Au-Cu Alloys. Journal of Composites Science. 2022; 6(12):383. https://doi.org/10.3390/jcs6120383
Chicago/Turabian StyleTrong, Dung Nguyen, Van Cao Long, Umut Saraç, Van Duong Quoc, and Ştefan Ţălu. 2022. "First-Principles Calculations of Crystallographic and Electronic Structural Properties of Au-Cu Alloys" Journal of Composites Science 6, no. 12: 383. https://doi.org/10.3390/jcs6120383
APA StyleTrong, D. N., Long, V. C., Saraç, U., Quoc, V. D., & Ţălu, Ş. (2022). First-Principles Calculations of Crystallographic and Electronic Structural Properties of Au-Cu Alloys. Journal of Composites Science, 6(12), 383. https://doi.org/10.3390/jcs6120383