
Review on Characterization of Biochar Derived from Biomass Pyrolysis via Reactive Molecular Dynamics Simulations



Abstract
:1. Introduction
2. Fundamentals of ReaxFF MD Simulations
2.1. General Force Field
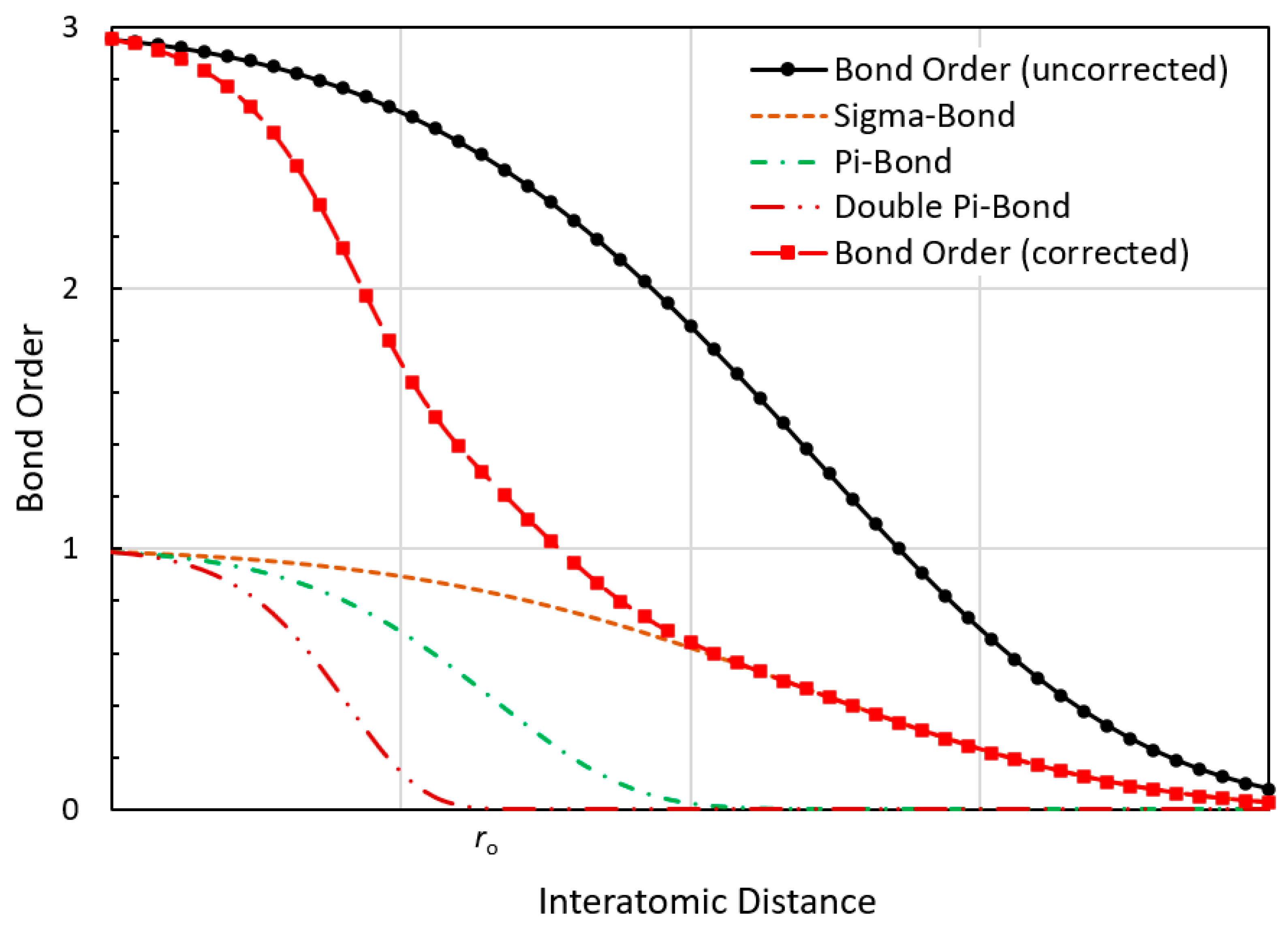
2.2. Bond Order and Bond Energy
2.3. Atomic Over-Coordination
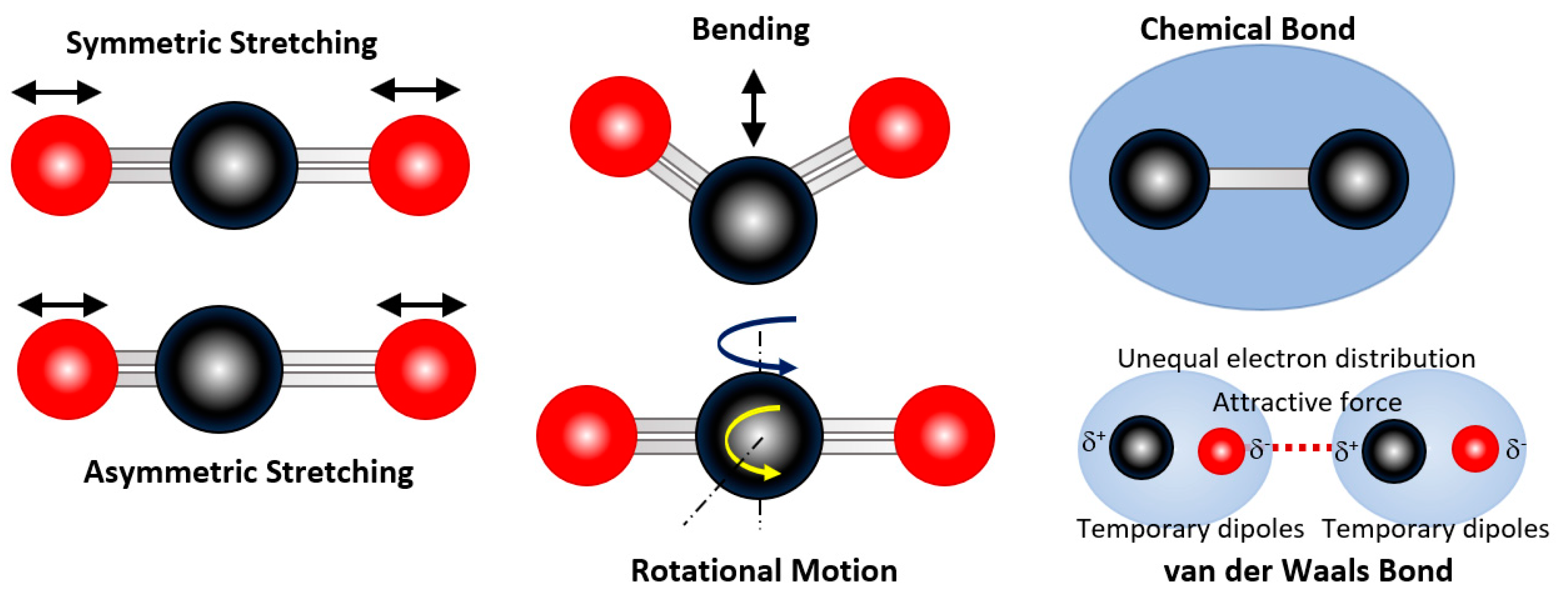
2.4. Valence Angle and Torsional Angle Interaction Terms
2.5. Coulombic and van der Waals Interaction Terms
2.6. Conjugated System Term
2.7. Force Field Optimization Procedure
3. Results and Discussion
3.1. The Compositions and Physicochemical Properties of Biomass Feedstocks and the Produced Biochar
3.2. Carbonization Reactions in Biomass Pyrolysis Processes
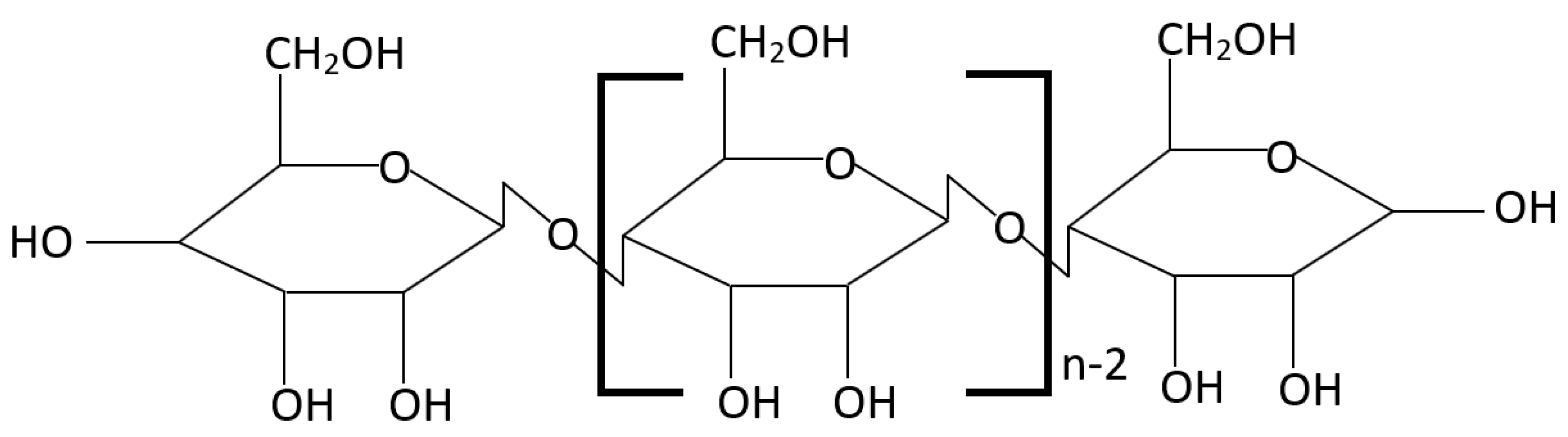
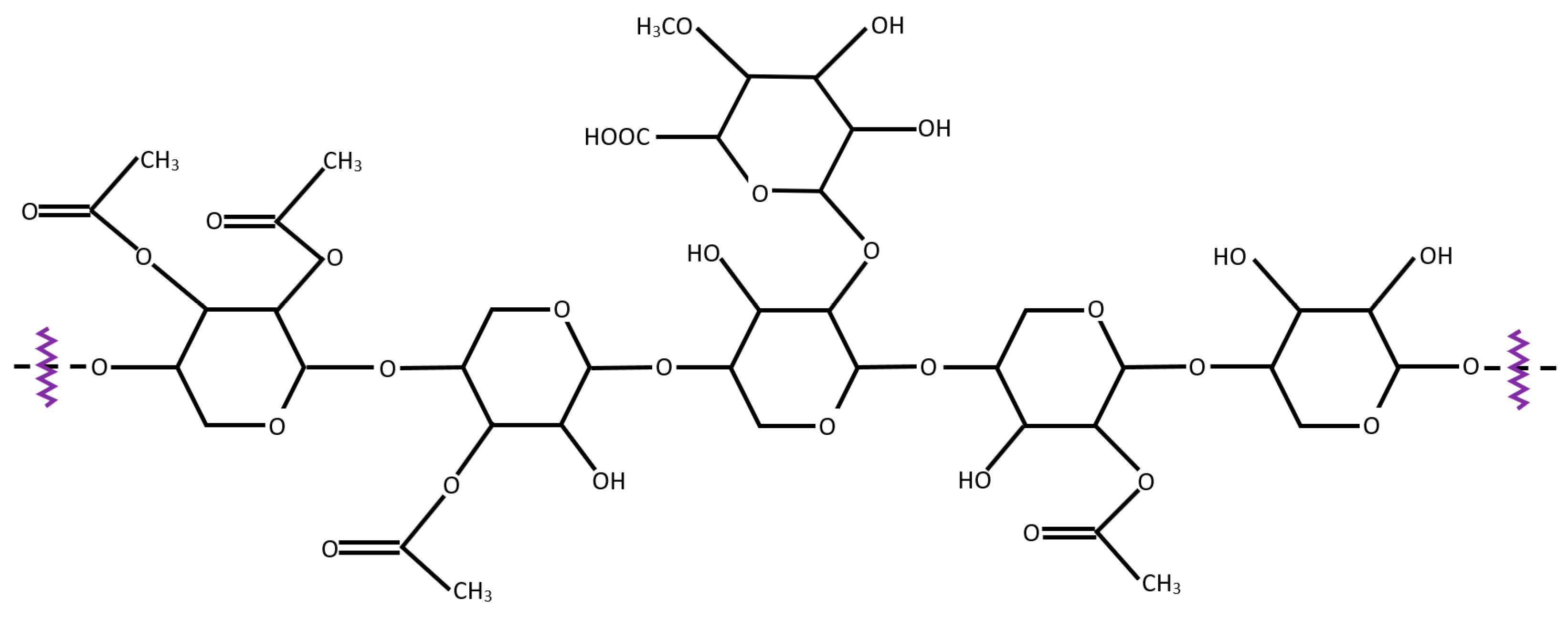
3.2.1. Reactivity of Cellulose in Pyrolysis
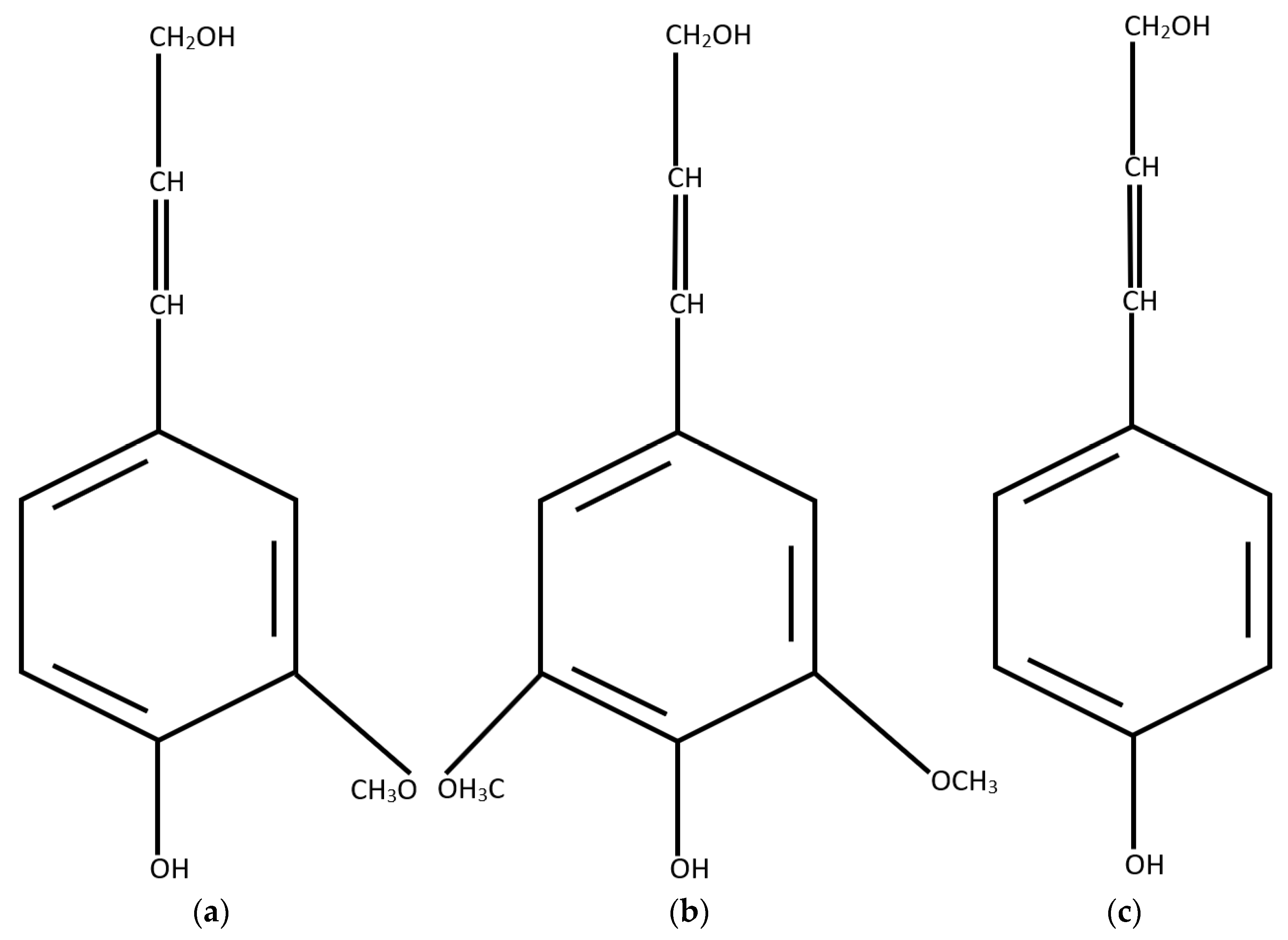
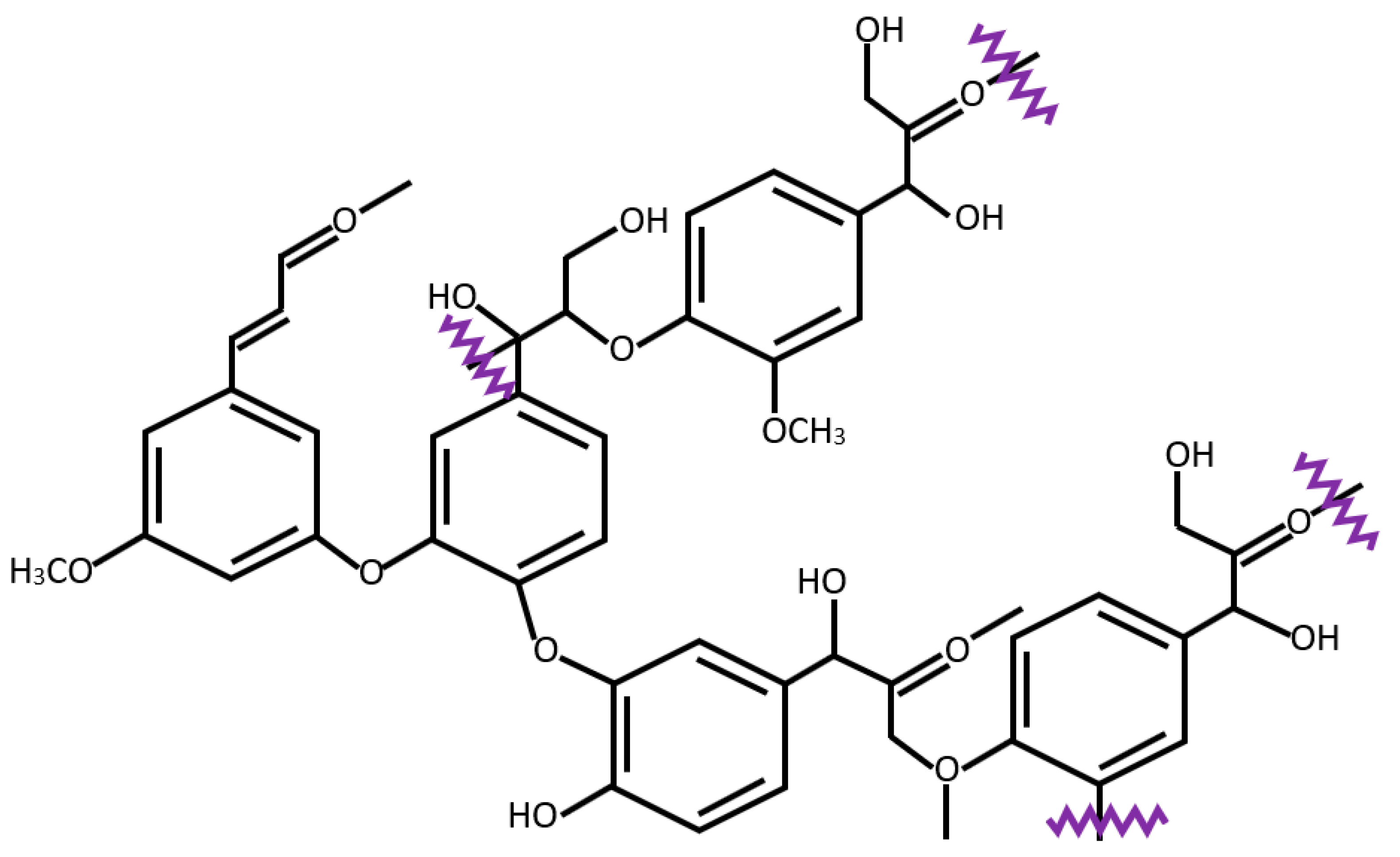
3.2.2. Reactivity of Lignin in Pyrolysis
3.2.3. Reactivity of Hemicellulose in Pyrolysis
3.3. Additional Notes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, F.; Cao, J.-P.; Zhao, X.-Y.; Ren, J.; Wang, J.-X.; Fan, X.; Wei, X.-Y. Nitrogen evolution during fast pyrolysis of sewage sludge under inert and reductive atmospheres. Energy Fuels 2017, 31, 7191–7196. [Google Scholar] [CrossRef]
- Sun, H.; Wu, C. Autothermal CaO looping biomass gasification for renewable syngas production. Environ. Sci. Technol. 2019, 53, 9298–9305. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, Y.-L.; Zhao, X.-Y.; Cao, J.-P. Biomass thermochemical conversion: A review on tar elimination from biomass catalytic gasification. J. Energy Inst. 2020, 93, 1083–1098. [Google Scholar] [CrossRef]
- Li, H.; Xu, B.; Jin, H.; Luo, K.; Fan, J. Molecular dynamics investigation on the lignin gasification in supercritical water. Fuel Process. Technol. 2019, 192, 203–209. [Google Scholar] [CrossRef]
- Marangwanda, G.T.; Madyira, D.M.; Babarinde, T.O. Combustion models for biomass: A review. Energy Rep. 2020, 6, 664–672. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Garcia, B.; Alves, O.; Rijo, B.; Lourinho, G.; Nobre, C. Biochar: Production, applications, and market prospects in Portugal. Environments 2022, 9, 95. [Google Scholar] [CrossRef]
- Gupta, R.; Dubey, M.; Kharel, P.; Gu, Z.; Fan, Q. Biochar activated by oxygen plasma for supercapacitors. J. Power Sources 2015, 274, 1300–1305. [Google Scholar] [CrossRef]
- Clough, T.J.; Condron, L.M.; Kammann, C.; Müller, C. A review of biochar and soil nitrogen dynamics. Agronomy 2013, 3, 275–293. [Google Scholar] [CrossRef]
- Steiner, C.; McLaughlin, H.; Harley, A.; Flora, G.; Larson, R.; Reed, A. United States Biochar Initiative 2010: U.S.-Focused Biochar Report–Assessment of Biochar’s Benefits for the United States of America; United States Center for Energy and Environmental Security; United States Biochar Initiative: Morgantown, WV, USA, 2010; p. 84. [Google Scholar]
- Kumar, A.; Singh, J.; Churasia, A. Production of biodiesel from soybean oil biomass as renewable energy source. J. Environ. Biol. 2016, 37, 1303–1307. [Google Scholar]
- Paletto, A.; Bernardi, S.; Pieratti, E.; Teston, F.; Romagnoli, M. Assessment of environmental impact of biomass power plants to increase the social acceptance of renewable energy technologies. Heliyon 2019, 5, e02070. [Google Scholar] [CrossRef]
- Hodges, D.G.; Chapagain, B.; Watcharaanantapong, P.; Poudyal, N.C.; Kline, K.L.; Dale, V.H. Opportunities and attitudes of private forest landowners in supplying woody biomass for renewable energy. Renew. Sustain. Energy Rev. 2019, 113, 109205. [Google Scholar] [CrossRef]
- Omoriyekomwan, J.E.; Tahmasebi, A.; Zhang, J.; Yu, J. Synthesis of Super-Long Carbon Nanotubes from Cellulosic Biomass under Microwave Radiation. Nanomaterials 2022, 12, 737. [Google Scholar] [CrossRef]
- Hersh, B.; Mirkouei, A.; Sessions, J.; Rezaie, B.; You, Y. A review and future directions on enhancing sustainability benefits across food-energy-water systems: The potential role of biochar-derived products. AIMS Environ. Sci. 2019, 6, 379–416. [Google Scholar] [CrossRef]
- Bartoli, M.; Giorcelli, M.; Jagdale, P.; Rovere, M.; Tagliaferro, A. A review of non-soil biochar applications. Materials 2020, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Allohverdi, T.; Mohanty, A.K.; Roy, P.; Misra, M. A review on current status of biochar uses in agriculture. Molecules 2021, 26, 5584. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, D.; Celesti, C.; Espro, C.; Ferlazzo, A.; Giofrè, S.V.; Scuderi, M.; Scalese, S.; Gabriele, B.; Mancuso, R.; Ziccarelli, I.; et al. Orange-peel-derived nanobiochar for targeted cancer therapy. Pharmaceutics 2022, 14, 2249. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.E.; Ledesma, B.; Román, S.; Bonelli, P.R.; Cukierman, A.L. Development and characterization of activated hydrochars from orange peels as potential adsorbents for emerging organic contaminants. Bioresour. Technol. 2015, 183, 221–228. [Google Scholar] [CrossRef]
- Wang, T.; Liu, J.; Zhang, Y.; Zhang, H.; Chen, W.-Y.; Norris, P.; Pan, W.-P. Use of a non-thermal plasma technique to increase the number of chlorine active sites on biochar for improved mercury removal. Chem. Eng. J. 2018, 331, 536–544. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Preparation, Modification and Environmental Application of Biochar: A Review. J. Clean. Prod. 2019, 227, 1002–1022. [Google Scholar] [CrossRef]
- Adhamash, E.; Pathak, R.; Qiao, Q.; Zhou, Y.; McTaggart, R. Gamma-radiated biochar carbon for improved supercapacitor performance. RSC Adv. 2020, 10, 29910–29917. [Google Scholar] [CrossRef]
- Alam, M.S.; Alessi, D.S. Chapter 4—Modeling the Surface Chemistry of Biochars. In Biochar from Biomass and Waste—Fundamentals and Applications; Ok, Y.S., Tsang, D.C.W., Bolan, N., Novak, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 59–72. [Google Scholar]
- Mukherjee, A.; Zimmerman, A.R.; Harris, W. Surface chemistry variations among a series of laboratory-produced biochars. Geodema 2011, 163, 247–255. [Google Scholar] [CrossRef]
- Das, C.; Tamrakar, S.; Kiziltas, A.; Xie, X. Incorporation of biochar to improve mechancal, thermal and electrical properties of polymer composites. Polymers 2021, 13, 2663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Duan, W.; Peng, H.; Pan, B.; Xing, B. Functional biochar and its balanced design. ACS Anvironmental 2022, 2, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Nguyen, N.-T.; Le, T.-T.; Duong, C.-C.; Duong, T.-T. Specifically designed amine functional group doped sludge biochar for inorganic and organic arsenic removal. Sustain. Environ. Res. 2021, 31, 28. [Google Scholar] [CrossRef]
- Bamdad, H.; Papari, S.; MacQuarrie, S.; Hawboldt, K. Study of surface heterogeneity and nitrogen functinalization of biochars: Molecular modeling approach. Carbon 2021, 171, 161–170. [Google Scholar] [CrossRef]
- Hong, D.; Gao, P.; Wang, C. A comprehensive understanding of the synergistic effect during co-pyrolysis of polyvinyl chloride (PVC) and coal. Energy 2022, 239, 122258. [Google Scholar] [CrossRef]
- Wang, M.; Gao, J.; Xu, J.; Du, Q.; Zhang, Y. Effect of H2O on the transformation of sulfur during demineralized coal pyrolysis: Molecular dynamics simulation using ReaxFF. Energy Fuels 2021, 35, 2379–2390. [Google Scholar] [CrossRef]
- Cui, L.; Fan, Q.; Sun, J.; Quan, G.; Yan, J.; Hina, K.; Wang, H.; Zhang, Z.; Hussain, Q. Change in surface characteristics and adsorption properties of 2, 4, 6-trichlorophenol following Fenton-like aging of biochar. Sci. Rep. 2021, 11, 4293. [Google Scholar] [CrossRef]
- Muretta, J.E. Investigation of the Structure, Chemistry, and Functional Performance of Novel Biochars. Ph.D. Dissertation, Montana Technological University, Butte, MT, USA, 2022. [Google Scholar]
- Feng, D.; Guo, D.; Zhang, Y.; Sun, S.; Zhao, Y.; Shang, Q.; Sun, H.; Wu, J.; Tan, H. Functionalized construction of biochar with hierarchical pore strucyures and surface O-/N-containing groups for phenol adsorption. Chem. Eng. J. 2021, 410, 127707. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, B. A direct observation of the fine aromatic clusters and molecular structures of biochars. Environ. Sci. Technol. 2017, 61, 5473–5482. [Google Scholar] [CrossRef]
- Jimenez-Cordero, D.; Heras, F.; Alonso-Morales, N.; Gilarranz, M.A.; Rodriguez, J.J. Porous structure and morphology of granular chars from flash and conventional pyrolysis of grape seeds. Biomass Bioenergy 2013, 54, 123–132. [Google Scholar] [CrossRef]
- Brewer, C.E.; Chuang, V.J.; Masiella, C.A.; Gonnermann, H.; Gao, X.; Dugan, B.; Driver, L.E.; Panzacchi, P.; Zygourakis, K.; Davies, C.A. New approaches to measuring biochar density and porosity. Biomass Bioenergy 2014, 66, 176–185. [Google Scholar] [CrossRef]
- Vidal-Vidal, Á.; Faza, O.N.; López, C.S. CO2 complexes with five-membered heterocycles: Structure, topology, and spectroscopic characterization. J. Phys. Chem. A 2017, 121, 9118–9130. [Google Scholar] [CrossRef] [PubMed]
- Yaashikaa, P.R.; Kumar, P.S.; Varijani, S.; Saravanan, A. A critical review on the biochar production techniques, characterizaiton, stability and applications for circular bioeconomy. Biotechnol. Rep. 2020, 28, e00570. [Google Scholar] [CrossRef]
- Keiluwrit, M.; Nico, P.S.; Johnson, M.G.; Kleber, M. Dynamic molecular structure of plant biomass-derived black carbon (biochar). Environ. Sci. Technol. 2010, 44, 1247–1253. [Google Scholar] [CrossRef]
- Kaal, J.; Cortizas, A.M.; Reyes, O.; Soliño, M. Molecular characterization of Ulex europaeus biochar obtained from laboratory heat treatment experiments—A pyrolysis—GC/MS study. J. Anal. Appl. Pyrolysis 2012, 95, 205–212. [Google Scholar] [CrossRef]
- Conte, P.; Bertani, R.; Sgarbossa, P.; Bambina, P.; Schmidt, H.-P.; Raga, R.; Papa, G.L.; Martino, D.F.C.; Meo, P.L. Recent developments in understanding biochar’s pysical-chemistry. Agronomy 2021, 11, 615. [Google Scholar] [CrossRef]
- Zuhara, S.; Mackey, H.R.; Al-Ansari, T.; Mckay, G. A review of propsects and current scenarios of biomass co-pyrolysis for water treatment. Biomass Convers. Biorefinery 2022. [Google Scholar] [CrossRef]
- Mehdi, R.; Khoja, A.H.; Naqvi, S.R.; Gao, N.; Amin, N.A.S. A review on production and surface modifications of biochar materials via biomass pyrolysis process for supercapacitor applications. Catalysts 2022, 12, 798. [Google Scholar] [CrossRef]
- Najmi, L.; Hu, Z. Review on molecular dynamics simulations of effects of carbon nanotubes (CNTs) on electrical and thermal conductivities of CNT-modified polymeric composites. J. Compos. Sci. 2023, 7, 165. [Google Scholar] [CrossRef]
- Zhao, N.; Lv, Y.; Yang, X. A new 3D conceptual structure modeling of biochars by molecular mechanic and molecular dynamic simulation. J. Solids Sediments 2017, 17, 641–655. [Google Scholar] [CrossRef]
- Dash, B. Carbon dioxide capture by nitrogen containing organic materials—A density functional theory investigation. Comput. Theor. Chem. 2018, 1128, 1–14. [Google Scholar] [CrossRef]
- Mrozik, W.; Minofar, B.; Thongsamer, T.; Wiriyaphong, N.; Khawkomol, S.; Plaimart, J.; Vakros, J.; Karapanagioti, H.; Vinitnantharat, S.; Werner, D. Valorisation of agricultural waste derived biochars in aquaculture to remove organic micropollutants from water—Experimental study and molecular dynamics simulations. J. Environ. Manag. 2021, 300, 113717. [Google Scholar] [CrossRef] [PubMed]
- Ouachtak, H.; Guerdaoui, A.E.; Haouti, R.E.; Haounati, R.; Ighnih, H.; Toubi, Y.; Alakhras, F.; Rehman, R.; Hafid, N.; Addi, A.A.; et al. Combined molecular dynamics simulations and experimental studies of the removal of cationic dyes on the eco-friendly adsorbent of activated carbon decorated montmorillonite Mt@AC. RSC Adv. 2023, 13, 5027–5044. [Google Scholar] [CrossRef]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. A review of multiscale computational methods in polymeric materials. Polymers 2017, 9, 16. [Google Scholar] [CrossRef]
- Mao, Q.; Feng, M.; Jiang, X.Z.; Ren, Y.; Luo, K.H.; van Duin, A.C.T. Classical and reactive molecular dyanmics: Principles and applications in combustion and energy systems. Prog. Energy Combust. Sci. 2023, 97, 101084. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard III, W.A. ReaxFF: A reactive force filed for hydrocarbons. J. Physcal Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Chenoweth, K.; van Duin, A.C.T.; Goddard III, W.A. ReaxFF force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Russo, M.F., Jr.; van Duin, A.C.T. Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering. Nucl. Instrum. Methods Phys. Res. B 2011, 269, 1549–1554. [Google Scholar] [CrossRef]
- Aktulga, H.M.; Pandit, S.A.; van Duin, A.C.T.; Grama, A.Y. Reactive molecular dynamics: Numerical methods and algorithmic techniques. SIAM J. Sci. Comput. 2012, 34, C1–C23. [Google Scholar] [CrossRef]
- Beste, A. ReaxFF study of the oxidation of softwood lignin in view of carbon fiber production. Energy Fuels 2014, 28, 7007–7013. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. NPJ Comput. Mater. 2016, 2, 15011. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, L.; Wang, J.; Lin, S. Reactive molecular dynamics simulations of biomass pyrolysis and combustion under various oxidative and humidity environments. Ind. Eng. Chem. Res. 2017, 56, 12278–12288. [Google Scholar] [CrossRef]
- Leven, I.; Hao, H.; Das, A.K.; Head-Gordon, T. A reactive force field with coarse-grained electrons for liquid water. J. Phys. Chem. Lett. 2020, 11, 9240–9247. [Google Scholar] [CrossRef]
- Liu, Z.; Ku, X.; Jin, H. Pyrolysis mechanism of wheat straw based on ReaxFF molecular dynamics simulations. ACS Omega 2022, 7, 21075–21085. [Google Scholar] [CrossRef]
- Friesner, R.A. Ab initio quantum chemistry: Methodology and applications. Proc. Natl. Acad. Sci. USA 2005, 102, 6648–6653. [Google Scholar] [CrossRef]
- Lee, H.M.; Youn, I.S.; Saleh, M.; Lee, J.W.; Kim, K.S. Interactions of CO2 with various functional molecules. Phys. Chem. Chem. Phys. 2015, 17, 10925. [Google Scholar] [CrossRef]
- Zhao, N.; Zhao, C.; Liu, K.; Zhang, W.; Tsang, D.C.W.; Yang, Z.; Yang, X.; Yan, B.; Morel, J.L.; Qiu, R. Experimental and DFT investigation on N-functionalized biochars for enhanced removel of Cr(VI). Environ. Pollut. 2021, 291, 118244. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef]
- Purse, M.; Holmes, B.; Sacchi, M.; Howlin, B. Simulation the complete pyrolysis and charring process of phenol-formaldehyde resins using reactive molecular dynamics. J. Mater. Sci. 2022, 57, 7600–7620. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Damsté, J.S.S. Computational chemical investigation into isorenieratene cyclisation. Org. Geochem. 2003, 34, 515–526. [Google Scholar] [CrossRef]
- Chen, N.; Lusk, M.T.; van Duin, A.C.T.; Goddard III, W.A. Mechanical properties of connected carbon nanorings via molecular dynamics simulation. Phys. Rev. B 2005, 72, 085416. [Google Scholar] [CrossRef]
- Han, S.S.; Kang, J.K.; Lee, H.M.; van Duin, A.C.T.; Goddard III, W.A. Liquefaction of H2 molecules upon exterior surfaces of carbon nanotube bundles. Appl. Phys. Lett. 2005, 86, 203108. [Google Scholar] [CrossRef]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity-equalization method for the calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Janssens, G.O.A.; Baekelandt, B.G.; Toufar, H.; Mortier, W.J.; Schoonheydt, R.A. Comparison of cluster and infinite crystal calculations on zeolites with the electronegativity equalization method (EEM). J. Phys. Chem. 1995, 99, 3251–3258. [Google Scholar] [CrossRef]
- Rappe, A.K.; Goddard, W.A., III. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Johannes, T. Zur Kenntnis der ungesättigten verbindungen. Justus Liebig’s Ann. Der Chem. 1899, 306, 87–142. [Google Scholar]
- UPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; The “Gold Book”; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]
- March, J. Advanced Organic Chemistry Reactions, Mechanisms and Structure, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1985; ISBN 0-471-85472-7. [Google Scholar]
- Smith, J.G. Chapter 16: Conjugation, Resonance, and Dienes. In Organic Chemistry, 5th ed.; Iswar Chandra Vidyasagar College: Belonia, Tripura, India, 2022; pp. 604–640. ISBN 9780078021558. [Google Scholar]
- van Duin, A.C.T.; Baas, J.M.A.; van de Graaf, B.J. Delft molecular mechanics: A new approach to hydrocarbon force fields. Inclusion of a geometry-dependent charge calculation. J. Chem. Soc. Faraday Trans. 1994, 90, 2881–2895. [Google Scholar] [CrossRef]
- Li, G.; Zheng, F.; Huang, Q.; Wang, J.; Niu, B.; Zhang, Y.; Long, D. Molecular insight into pyrolysis processes via reactive force field molecular dynamics: A state-of-the-art review. J. Anal. Appl. Pyrolysis 2022, 166, 105620. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Wijitkosum, S.; Jiwnok, P. Elemental composition of biochar obtained from agricultural waste for soil amendment and carbon sequestration. Appl. Sci. 2019, 9, 3980. [Google Scholar] [CrossRef]
- Ippolito, J.A.; Cui, L.; Kammann, C.; Wrage-Mönnig, N.; Estavillo, J.M.; Fuertes-Mendizabal, T.; Cayuela, M.L.; Sigua, G.; Novak, J.; Spokas, K.; et al. Feedstock choice, pyrolysis temperature and type influence biochar characteristics: A comprehensive meta-data analysis review. Biochar 2020, 2, 421–438. [Google Scholar] [CrossRef]
- Libra, J.A.; Ro, K.S.; Kammann, C.; Funke, A.; Berge, N.D.; Neubauer, Y.; Titirici, M.-M.; Uhner, C.F.; Bens, O.; Kern, J.; et al. Hydrothermal carbonizationof biomass residuals: A comparative review of the chemistry, processes and applications of wet and dry pyrolysis. Biofuels 2011, 2, 71–106. [Google Scholar] [CrossRef]
- Brownsort, P.A. Biomass Pyrolysis Processes: Performance Parameters and Their Influence on Biochar System benefits. Master’s Dissertation, The University of Edinburgh, Edinburgh, Scotland, UK, 2009. [Google Scholar]
- Antal, M.J.; Grønli, M. The art, science, and technology of charcoal production. Ind. Eng. Chem. Res. 2003, 42, 1619–1640. [Google Scholar] [CrossRef]
- Lucian, M.; Fiori, L. Hydrothermal carbonization of waste biomass: Process design, modeling, energy efficiency and cost analysis. Energies 2017, 10, 211. [Google Scholar] [CrossRef]
- Arvelos, S.; Abrahão, O., Jr.; Hori, C.E. ReaxFF molucular dynamics study on the pyrolysis process of cyclohexanone. J. Analtical Appl. Pyrolysis 2019, 141, 104620. [Google Scholar] [CrossRef]
- Xu, F.; Liu, H.; Wang, Q.; Pan, S.; Zhao, D.; Liu, Q.; Liu, Y. ReaxFF-based molecular dyanamics simulation of the intial pyrolysis mechanism of lignite. Fuel Process. Technol. 2019, 195, 106147. [Google Scholar] [CrossRef]
- Xu, F.; Liu, H.; Wang, Q.; Pan, S.; Zhao, D.; Liu, Y. Study of non-isothermal pyrolysis mechanism of lignite using ReaxFF molecular dynamics simulations. Fuel 2019, 256, 115884. [Google Scholar] [CrossRef]
- Lele, A.; Kwon, H.; Ganeshan, K.; Xuan, Y.; van Duin, A.C.T. ReaxFF molecular dynamics study on pyrolysis of bicyclic compunds for aviation fuel. Fuel 2021, 297, 120724. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, Y. Reactive molecular dynamics simulation on degradation of tetracycline antibiotics treated by cold atmospheric plasmas. Molecules 2023, 28, 3850. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Li, X.; Guo, L. Pyrolysis simulations of Fugu coal by large-scale ReaxFF molecular dynamics. Fuel Process. Technol. 2018, 178, 197–205. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Guo, L. Investigation of N behavior during coal pyrolysis and oxidation using ReaxFF molecular dynamics. Fuel 2018, 233, 867–876. [Google Scholar] [CrossRef]
- Bhoi, S.; Banerjee, T.; Mohanty, K. Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF. Fuel 2014, 136, 326–333. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Liu, J.; Wang, Z.; Kong, B.; Gong, X.; Yang, X.; Lin, W.; Guo, L. Study of high density polyethylene (HDPE) pyrolysis with reactive molecular dynamics. Polym. Degrad. Stab. 2014, 104, 62–70. [Google Scholar] [CrossRef]
- Zhao, T.; Li, T.; Xin, Z.; Zou, L.; Zhang, L. A ReaxFF-based molecular dynamics simulation of the pyrolysis mechanism for polycarbonate. Energy Fuels 2018, 32, 2156–2162. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, S.; Lv, Y.; Hu, P.; Huang, Y.; Kong, M.; Li, G. Atomic-scale insight into the pyrolysis of polycarbonate by ReaxFF-based reactive molecular dynamics simulation. Fuel 2021, 287, 119484. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, Z.; Li, X.; Qiao, X.; Song, W.; Guo, L. Initial reaction mechanisms of cellulose pyrolysis revealed by ReaxFF molecular dynamics. Fuel 2016, 177, 130–141. [Google Scholar] [CrossRef]
- Diao, Z.; Zhao, Y.; Chen, B.; Duan, C.; Song, S. ReaxFF reactive force field for molecular dynamics simulations of epoxy resin thermal decomposition with model compound. J. Anal. Appl. Pyrolysis 2013, 104, 618–624. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Y.; Chen, X.; Wen, H.; Xiao, S. Theoretical study on decomposition mechanism of insulating epoxy resin cured by anhydride. Polymers 2017, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, P.; Luo, W.; Zhang, J.; Yu, H.; Chen, F.; Zhang, F. Simulation of pyrolysis of crosslinked epoxy resin using ReaxFF molecular dynamics. Comput. Theor. Chem. 2021, 1200, 113240. [Google Scholar] [CrossRef]
- Natali, M.; Kenny, J.M.; Torre, L. Science and technology of polymeric ablative materials for thermal protection systems and propulsion devices: A review. Prog. Mater. Sci. 2016, 84, 192–275. [Google Scholar] [CrossRef]
- Castro-Marcano, F.; Kamat, A.M.; Russo, M.F., Jr.; van Duin, A.C.T.; Mathews, J.P. Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field. Combust. Flame 2012, 159, 1272–1285. [Google Scholar] [CrossRef]
- Odegard, G.M.; Jensen, B.D.; Gowtham, S.; Wu, J.; He, J.; Zhang, Z. Predicting mechanical response of crosslinked epoxy using ReaxFF. Chem. Phys. Lett. 2014, 591, 175–178. [Google Scholar] [CrossRef]
- Wang, S.; Guo, X.; Liang, T.; Zhou, Y.; Luo, Z. Mechanism research on cellulose pyrolysis by Py-GC/MS and subsequent density functional theory studies. Bioresour. Technol. 2012, 104, 722–728. [Google Scholar] [CrossRef]
- Paajamen, A.; Vaari, J. High-temperature decomposition of the cellulose molecule: A stochastic molecular dynamics study. Cellulose 2017, 24, 2713–2725. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, S.; Liao, Y.; Cen, K. Mechanism study of cellulose rapid pyrolysis. Ind. Eng. Chem. Res. 2004, 43, 5605–5610. [Google Scholar] [CrossRef]
- Davin, L.B.; Lewis, N.G. Lignin primary structures and dirigent sites. Curr. Opin. Biotechnol. 2005, 16, 407–415. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Guo, L.; Guo, X. Reaction mechanisms in pyrolysis of hardwood, softwood, and kraft lignin revealed by ReaxFF MD simulations. Energy Fuels 2019, 33, 11210–11225. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Guo, L. Initial reactivity of linkages and monomer rings in lignin pyrolysis revealed by ReaxFF molecular dynamics. Langmuir 2017, 33, 11646–11657. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.D.; Monti, S.; Barcaro, G.; Zhang, Z.; Barta, K. New mechanistic insights into the lignin β-O-4 linkage acidolysis with ethylene glycol stabilization aided by multilevel computational chemistry. ACS Sustain. Chem. Eng. 2021, 9, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Understanding Biomass Chemistry Using Multiscale Molecular Modeling Approach. In Catalysis for Clean Energy and Environmental Sustainability; Pant, K.K., Gupta, S.K., Ahmed, E., Eds.; Springer: Basel, Switzerland, 2021. [Google Scholar]
- Guidicianni, P.; Gargiulo, V.; Alfè, M.; Ragucci, R.; Ferreiro, A.I.; Rabaҫal, M.; Costa, M. Slow pyrolysis of xylan as pentose model compound for hardwood hemicellulose: A study of the catalytic effect of Na ions. J. Anal. Appl. Pyrolysis 2019, 137, 266–275. [Google Scholar] [CrossRef]
- Gargiulo, V.; Giudicianni, P.; Alfè, M.; Ragucci, R. About the influence of doping approach on the alkali metal catalyzed sloe pyrolysis of xylan. J. Chem. 2019, 2019, 9392571. [Google Scholar] [CrossRef]
- Gargiulo, V.; Ferreiro, A.I.; Giudicianni, P.; Tomaselli, S.; Costa, M.; Ragucci, R.; Alfe, M. Insights about the effect of composition, branching and molecular weight on the slow pyrolysis of xylose-based polysaccharides. J. Anal. Appl. Pyrolysis 2022, 161, 105369. [Google Scholar] [CrossRef]
- Shen, D.K.; Gu, S.; Bridgwater, A.V. Study on the pyrolytic behavior of xylan-based hemicellulose using TG-FTIR and Py-GC-FTIR. J. Anal. Appl. Pyrolysis 2010, 87, 199–206. [Google Scholar] [CrossRef]
- Chen, D.; Can, K.; Zhuang, X.; Gan, Z.; Zhou, J. Insight into biomass pyrolysis mechanism based on cellulose, hemicellulose, and lignin: Evolution of volatiles and kinetics, elucidation of reaction pathways, and characterization of gas, biochar and bio-oil. Combust. Flame 2022, 242, 112142. [Google Scholar] [CrossRef]
- Qu, T.; Guo, W.; Shen, L.; Xiao, J.; Zhao, K. Experimental study of biomass pyrolysis based on three major components: Hemicellulose, cellulose, and lignin. Ind. Eng. Chem. Res. 2011, 50, 10424–10433. [Google Scholar] [CrossRef]
- Zhou, X.; Li, W.; Mabon, R.; Broadbelt, L.J. A critical review on hemicellulose pyrolysis. Energy Technol. 2017, 5, 52–79. [Google Scholar] [CrossRef]
- Usino, D.O.; Supriyanto, P.Y.; Pettersson, A.; Richards, T. Influence of temperature and time on initial pyrolysis of cellulose and xylan. J. Anal. Appl. Pyrolysis 2020, 147, 104782. [Google Scholar] [CrossRef]
- Gao, Z.; Li, N.; Wang, Y.; Niu, W.; Yi, W. Pyrolysis behavior of xylan-based hemi-cellulose in a fixed bed reactor. J. Anal. Appl. Pyrolysis 2020, 146, 104772. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Product distribution from the fast pyrolysis of hemicellulose. ChemSusChem 2011, 4, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ku, X.; Jin, H.; Yang, S. Research on the microscopic reaction mechanism of cellulose pyrolysis using the molecular dynamics simulation. J. Anal. Appl. Pyrolysis 2021, 159, 105333. [Google Scholar] [CrossRef]
- Nimz, H. Beech lignin—Proposal of a constitutional scheme. Angew. Chem. Int. Ed. 1974, 13, 313–321. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyrolysis Type | SSA (m2/g) | CEC (cmol/kg) | AEC (cmol/kg) | CCE (%) | PV (m3/t) | APS (nm) | Ash (%) | pH | EC (dS/m) |
|---|---|---|---|---|---|---|---|---|---|
| Fast | 183 ± 17.3 | 44.9 ± 3.62 | 4.90 ± 3.45 | 6.10 ± 1.12 | 2.04 ± 0.81 | 52.3 ± 40.2 | 19.2 ± 19.2 | 8.7 ± 0.1 | 4.43 ± 0.50 |
| Slow | 98.6 ± 3.53 | 48.1 ± 3.12 | 5.33 ± 1.51 | 11.2 ± 0.98 | 3.66 ± 1.27 | 1190 ± 565 | 22.0 ± 0.51 | 8.7 ± 0.0 | 5.85 ± 1.58 |
| Feedstock source | SSA (m2/g) | CEC (cmol/kg) | AEC (cmol/kg) | CCE (%) | PV (m3/t) | APS (nm) | Ash (%) | pH | EC (dS/m) |
| Wood based | 184 ± 11.4 | 23.9 ± 1.87 | 5.65 ± 1.80 | 9.04 ± 1.17 | 7.01 ± 3.07 | 74.6 ± 44.4 | 10.2 ± 0.43 | 8.3 ± 0.1 | 6.20 ± 2.85 |
| Crop wastes | 98.2 ± 5.45 | 56.3 ± 3.92 | 4.51 ± 1.96 | 6.12 ± 0.97 | 2.05 ± 0.91 | 2320 ± 1150 | 21.1 ± 0.54 | 8.9 ± 0.1 | 5.72 ± 0.67 |
| Other grasses | 63.4 ± 8.84 | 63.316.4 | 2.05 ± 1.05 | — | 3.36 ± 3.30 | 268 ± 125 | 18.0 ± 1.01 | 8.9 ± 0.1 | 5.20 ± 0.93 |
| Manures/biosolids | 52.2 ± 4.23 | 66.1 ± 8.00 | 7.77 ± 7.52 | 14.2 ± 1.56 | 0.82 ± 0.30 | 27.3 ± 12.5 | 44.6 ± 0.97 | 8.9 ± 0.1 | 3.98 ± 0.41 |
| Pyrolysis Type | C (wt.%) | H (wt.%) | O (wt.%) | N (wt.%) | S (wt.%) |
|---|---|---|---|---|---|
| Fast | 60.6 ± 0.47 | 3.37 ± 0.08 | 19.1 ± 0.38 | 1.63 ± 0.06 | 0.085 ± 0.009 |
| Slow | 60.8 ± 0.34 | 3.36 ± 0.09 | 18.4 ± 0.29 | 1.63 ± 0.04 | 0.055 ± 0.004 |
| Feedstock source | C (wt.%) | H (wt.%) | O (wt.%) | N (wt.%) | S (wt.%) |
| Wood based | 70.5 ± 0.39 | 3.38 ± 0.08 | 17.7 ± 0.35 | 0.95 ± 0.03 | 0.044 ± 0.007 |
| Crop wastes | 61.4 ± 0.41 | 3.28 ± 0.10 | 18.1 ± 0.38 | 1.54 ± 0.06 | 0.039 ± 0.006 |
| Other grasses | 63.6 ± 0.72 | 5.11 ± 0.50 | 20.9 ± 0.74 | 1.80 ± 0.14 | 0.051 ± 0.021 |
| Manures/biosolids | 41.6 ± 0.68 | 2.73 ± 0.10 | 16.5 ± 0.70 | 2.42 ± 0.06 | 0.089 ± 0.006 |
| Corncobs/cassava rhizomes/cassava stems | 62.95–81.35 | 2.24–2.73 | 15.23–33.44 | 1.22–1.65 | — |
| Lignin Model | Species | Constituents | Number of C6H3 Units | ||
|---|---|---|---|---|---|
| H | G | S | |||
| Alder | Softwood | C6400H7200O2320 | 40 | 560 | 40 |
| Freudenberg | Softwood | C6980H7640O2280 | 200 | 480 | 40 |
| Nimz | Hardwood | C10854H11940O4062 | 42 | 612 | 396 |
| Marton | Kraft lignin | C5080H5040O1640S40 | 120 | 440 | 0 |
| CO (%) | CO2 (%) | CH4 (%) | H2O (%) | |
|---|---|---|---|---|
| Cellulose | 48.29 | 47.04 | 53.33 | 83.96 |
| Lignin | 7.32 | 4.68 | 40.00 | 5.30 |
| Hemicellulose | 44.39 | 48.28 | 6.67 | 10.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Wei, L. Review on Characterization of Biochar Derived from Biomass Pyrolysis via Reactive Molecular Dynamics Simulations. J. Compos. Sci. 2023, 7, 354. https://doi.org/10.3390/jcs7090354
Hu Z, Wei L. Review on Characterization of Biochar Derived from Biomass Pyrolysis via Reactive Molecular Dynamics Simulations. Journal of Composites Science. 2023; 7(9):354. https://doi.org/10.3390/jcs7090354
Chicago/Turabian StyleHu, Zhong, and Lin Wei. 2023. "Review on Characterization of Biochar Derived from Biomass Pyrolysis via Reactive Molecular Dynamics Simulations" Journal of Composites Science 7, no. 9: 354. https://doi.org/10.3390/jcs7090354
APA StyleHu, Z., & Wei, L. (2023). Review on Characterization of Biochar Derived from Biomass Pyrolysis via Reactive Molecular Dynamics Simulations. Journal of Composites Science, 7(9), 354. https://doi.org/10.3390/jcs7090354