
Energetic and Entropic Motifs in Vesicle Morphogenesis in Amphiphilic Diblock Copolymer Solutions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Methods and Software
2.2. Data Analytics
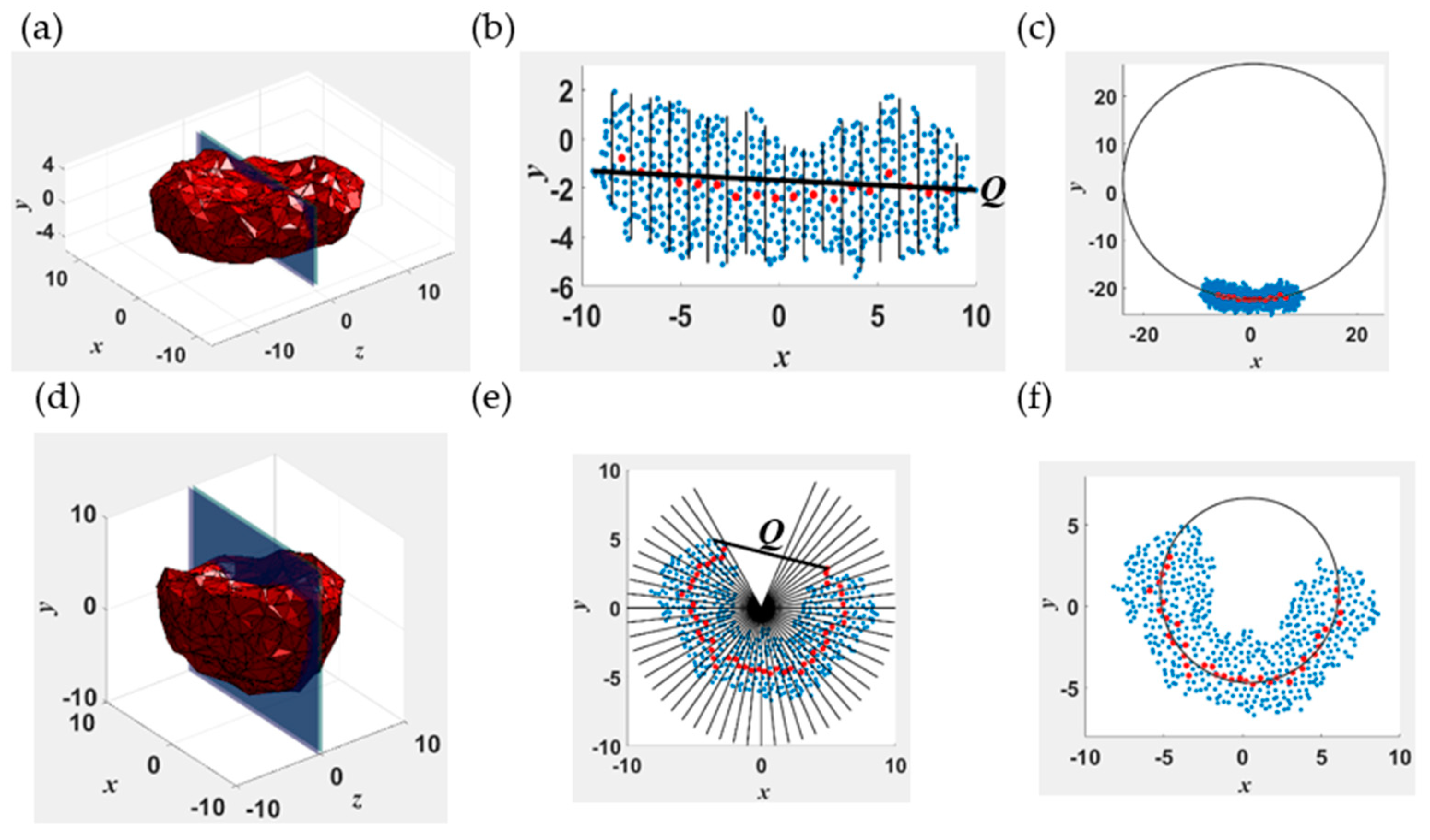
2.2.1. End-to-End Distance (Q) and Body Gaussian Curvature (KG)
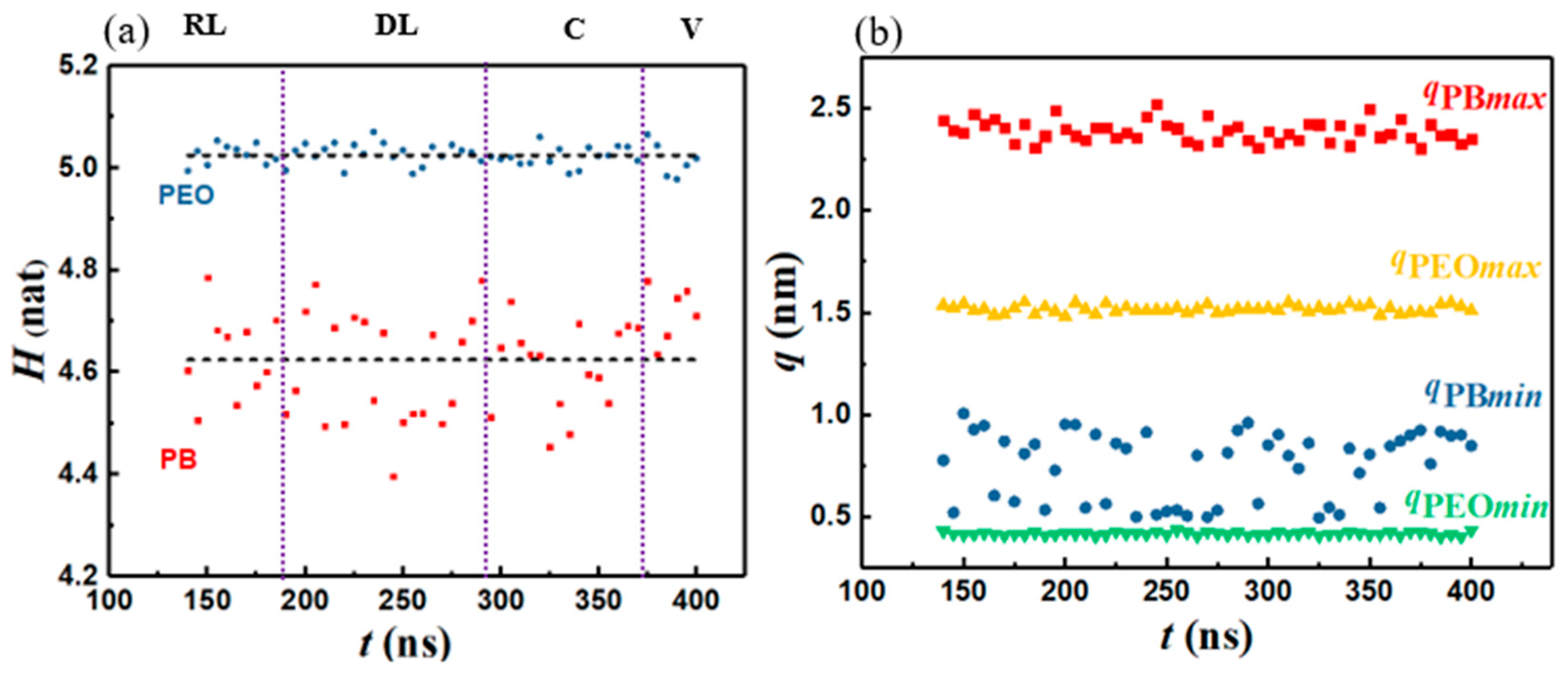
2.2.2. Information Entropy of Aggregates (H)
2.2.3. Pair Correlation Function (g(r))
3. Results and Discussion
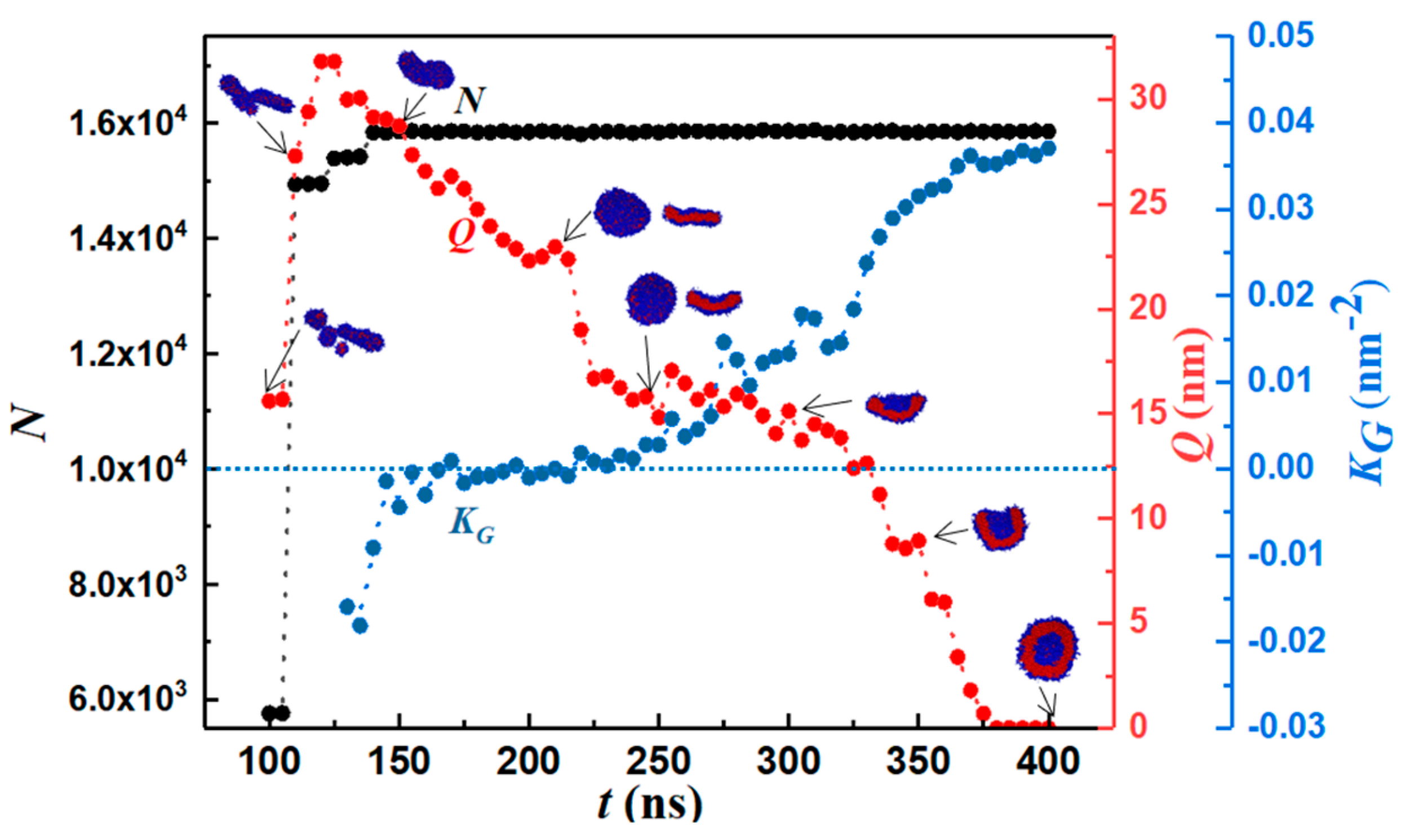
3.1. Morphology Evolution
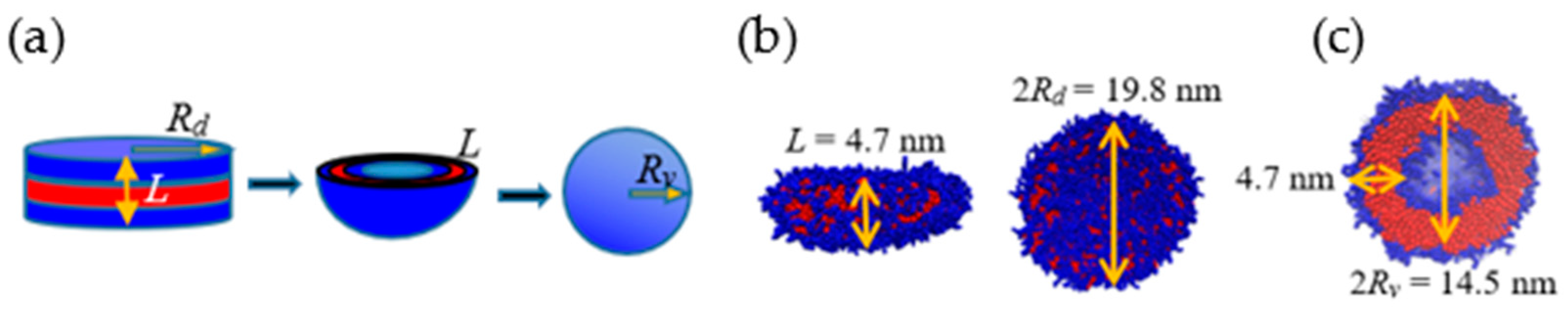
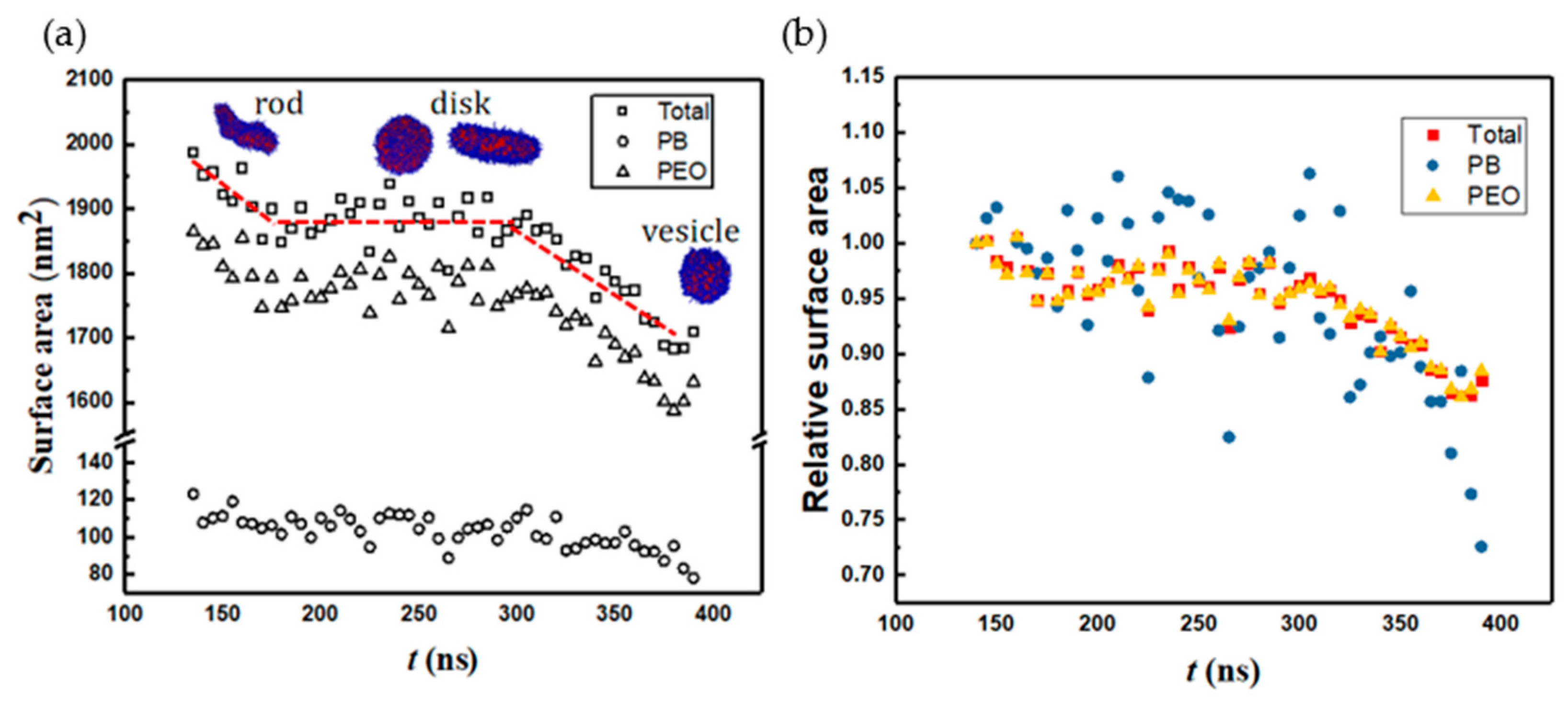
3.2. Prominent Intermediate Structures
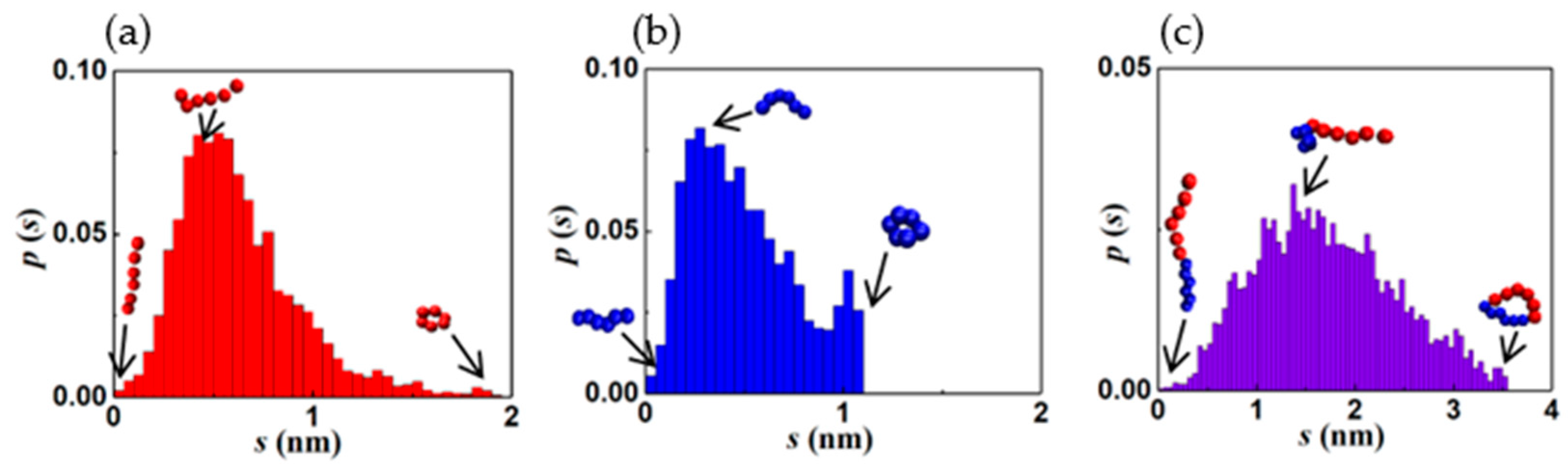
3.3. Entropic Motifs
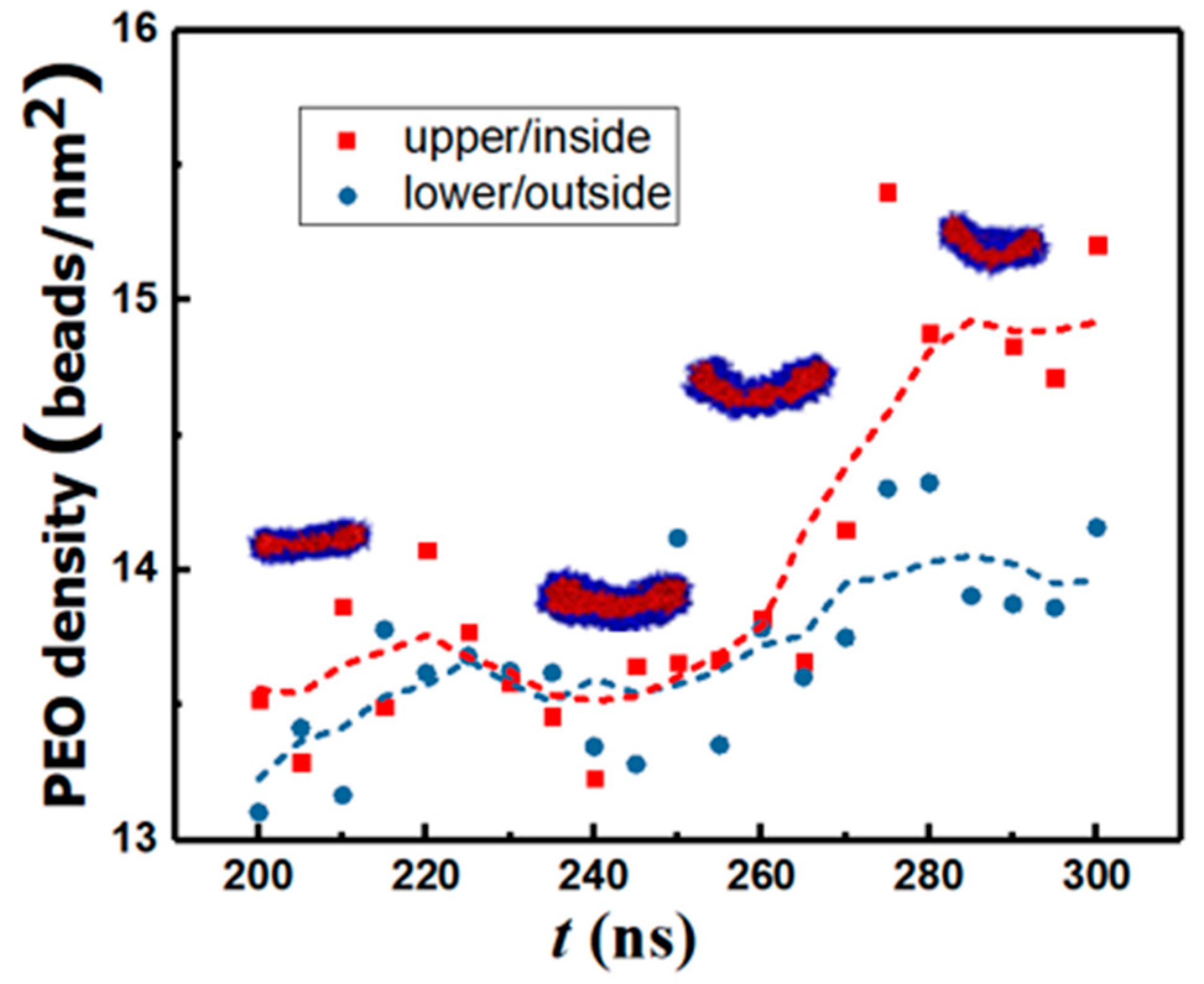
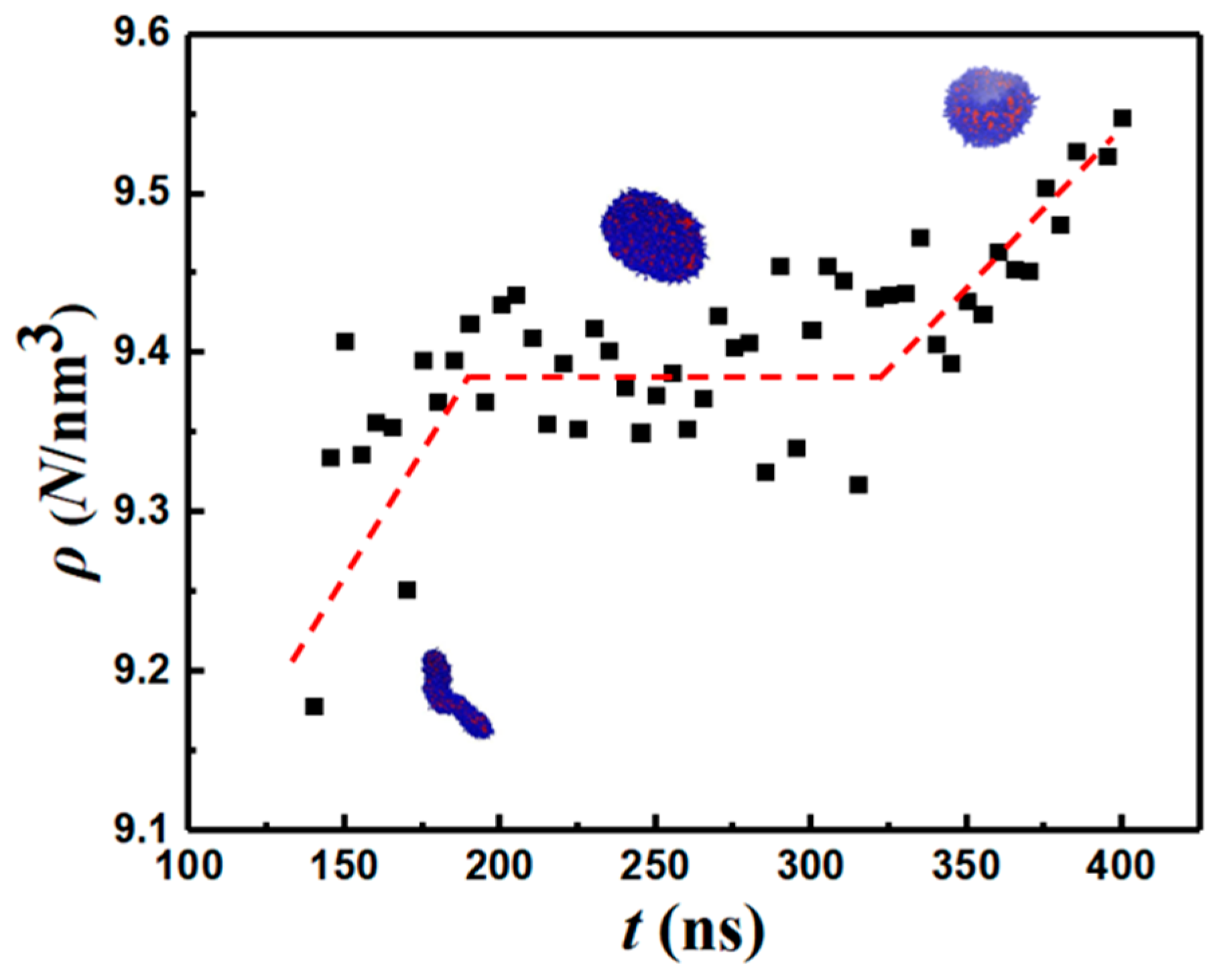
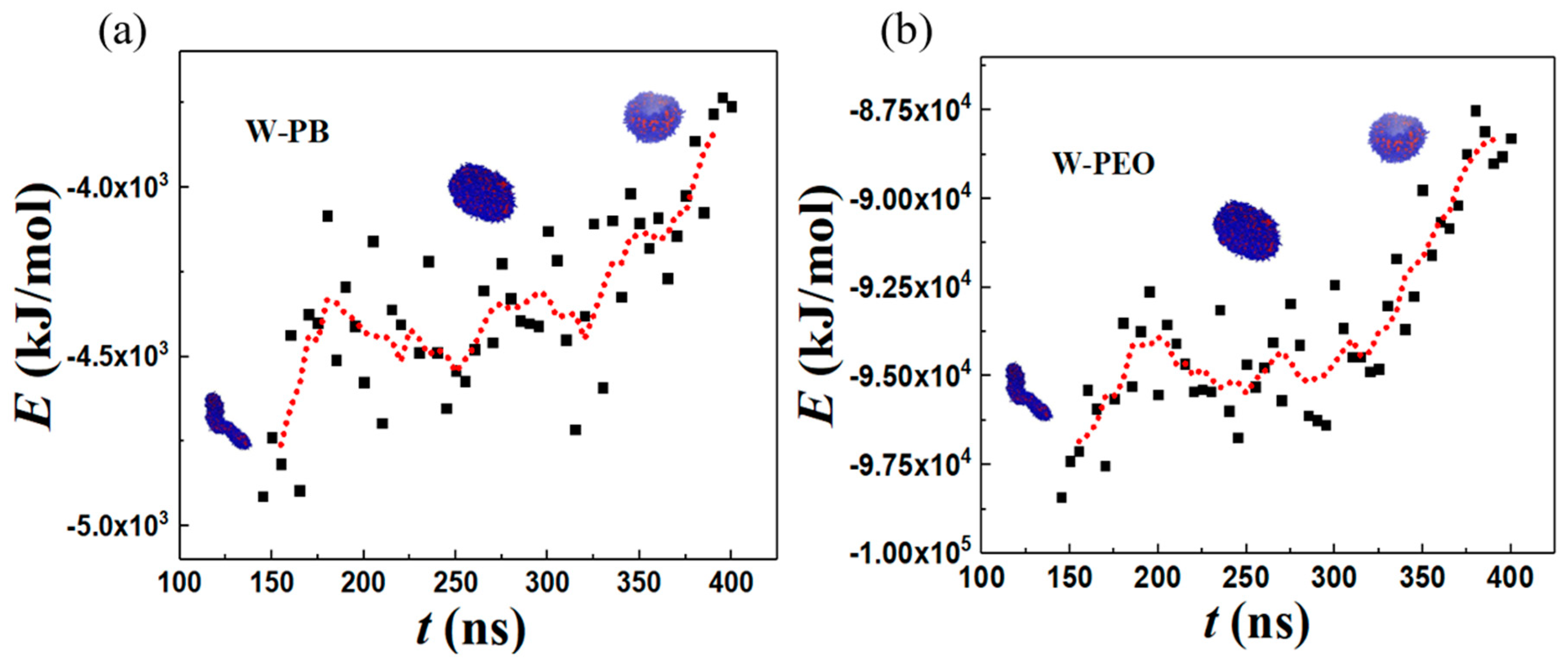
3.4. Energetic Motifs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Bleul, R.; Thiermann, R.; Maskos, M. Techniques to Control Polymersome Size. Macromolecules 2015, 48, 7396–7409. [Google Scholar] [CrossRef]
- Mohammadi, M.; Ramezani, M.; Abnous, K.; Alibolandi, M. Biocompatible Polymersomes-Based Cancer Theranostics: Towards Multifunctional Nanomedicine. Int. J. Pharm. 2017, 519, 287–303. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Zhang, P.-Y. Polymersomes in Nanomedicine—A Review. Curr. Nanosci. 2017, 13, 124–129. [Google Scholar] [CrossRef]
- Discher, D.E.; Ortiz, V.; Srinivas, G.; Klein, M.L.; Kim, Y.; Christian, D.; Cai, S.; Photos, P.; Ahmed, F. Emerging Applications of Polymersomes in Delivery: From Molecular Dynamics to Shrinkage of Tumors. Prog. Polym. Sci. 2007, 32, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Burkett, S.L.; Davis, M.E. Mechanism of Structure Direction In the Synthesis of Pure-Silica Zeolites. 2. Hydrophobic Hydration and Structural Specificity. Chem. Mater. 1995, 7, 1453–1463. [Google Scholar] [CrossRef]
- Deng, Y.; Wei, J.; Sun, Z.; Zhao, D. Large-Pore Ordered Mesoporous Materials Templated from Non-Pluronic Amphiphilic Block Copolymers. Chem. Soc. Rev. 2013, 42, 4054–4070. [Google Scholar] [CrossRef] [PubMed]
- Van Hest, J.C.; Delnoye, D.A.; Baars, M.W.; van Genderen, M.H.; Meijer, E.W. Polystyrene-Dendrimer Amphiphilic Block Copolymers with a Generation-Dependent Aggregation. Science 1995, 268, 1592–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Eisenberg, A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-Poly(Acrylic Acid) Block Copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef]
- Discher, B.M.; Won, Y.-Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough Vesicles Made From Diblock Copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef]
- Mai, Y.; Eisenberg, A. Self-assembly of Block Copolymers. Chem. Soc. Rev. 2012, 41, 5969. [Google Scholar] [CrossRef]
- Jain, S.; Bates, F.S. On the Origins of Morphological Complexity in Block Copolymer Surfactants. Science 2003, 300, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Won, Y.-Y.; Brannan, A.K.; Davis, H.T.; Bates, F.S. Cryogenic Transmission Electron Microscopy (Cryo-Tem) of Micelles and Vesicles Formed in Water by Poly(Ethylene Oxide)-Based Block Copolymers. J. Phys. Chem. B 2002, 106, 3354–3364. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Karayianni, M.; Pispas, S. Block Copolymer Solution Self-Assembly: Recent Advances, Emerging Trends, and Applications. J. Polym. Sci. 2021, 59, 1874–1898. [Google Scholar] [CrossRef]
- Li, S.; Byrne, B.; Welsh, J.E.; Palmer, A.F. Self-Assembled Poly(Butadiene)-b-Poly(Ethylene Oxide) Polymersomes as Paclitaxel Carriers. Biotechnol. Prog. 2007, 23, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Eisenberg, A.; Mrsic, J.; Maysinger, D. PCL-b-PEO Micelles as a Delivery Vehicle for Fk506: Assessment of a Functional Recovery of Crushed Peripheral Nerve. Drug Deliv. 2000, 7, 139–145. [Google Scholar] [CrossRef]
- Allen, C.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-Poly(Ethylene Oxide) Block Copolymer Micelles as a Novel Drug Delivery Vehicle for Neurotrophic Agents Fk506 and l-685,818. Bioconjugate Chem. 1998, 9, 564–572. [Google Scholar] [CrossRef]
- Boucher-Jacobs, C.; Rabnawaz, M.; Katz, J.S.; Even, R.; Guironnet, D. Encapsulation of catalyst in block copolymer micelles for the polymerization of ethylene in aqueous medium. Nat. Commun. 2018, 9, 841. [Google Scholar] [CrossRef]
- Cuomo, F.; Ceglie, A.; De Leonardis, A.; Lopez, F. Polymer Capsules for Enzymatic Catalysis in Confined Environments. Catalysts 2018, 9, 1. [Google Scholar] [CrossRef]
- Peters, R.J.; Marguet, M.; Marais, S.; Fraaije, M.W.; van Hest, J.C.; Lecommandoux, S. Cascade Reactions in Multicompartmentalized Polymersomes. Angew. Chem. Int. Ed. 2013, 53, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.A.; Nolte, R.J.; van Hest, J.C. Autonomous Movement of Platinum-Loaded Stomatocytes. Nat. Chem. 2012, 4, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Marguet, M.; Bonduelle, C.; Lecommandoux, S. Multicompartmentalized Polymeric Systems: Towards Biomimetic Cellular Structure and Function. Chem. Soc. Rev. 2013, 42, 512–529. [Google Scholar] [CrossRef]
- Che, H.; van Hest, J.C. Stimuli-Responsive Polymersomes and Nanoreactors. J. Mater. Chem. B 2016, 4, 4632–4647. [Google Scholar] [CrossRef]
- Jacobs, M.L.; Boyd, M.A.; Kamat, N.P. Diblock Copolymers Enhance Folding of a Mechanosensitive Membrane Protein during Cell-free Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 4031–4036. [Google Scholar] [CrossRef]
- Li, X.; Cooksey, T.J.; Kidd, B.E.; Robertson, M.L.; Madsen, L.A. Mapping Coexistence Phase Diagrams of Block Copolymer Micelles and Free Unimer Chains. Macromolecules 2018, 51, 8127–8135. [Google Scholar] [CrossRef]
- Holder, S.W.; Grant, S.C.; Mohammadigoushki, H. Nuclear Magnetic Resonance Diffusometry of Linear and Branched Wormlike Micelles. Langmuir 2021, 37, 3585–3596. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of Self-Assembly of Lipid Bilayers and Vesicles. Biochim. Biophys. Acta (BBA)-Biomembr. 1977, 470, 185–201. [Google Scholar] [CrossRef]
- Chen, L.; Shen, H.; Eisenberg, A. Kinetics and Mechanism of the Rod-to-Vesicle Transition of Block Copolymer Aggregates in Dilute Solution. J. Phys. Chem. B 1999, 103, 9488–9497. [Google Scholar] [CrossRef]
- Antonietti, M.; Förster, S. Vesicles and liposomes: A Self-Assembly Principle Beyond Lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar] [CrossRef]
- Liu, S.; Sureshkumar, R. Morphological Diversity in Diblock Copolymer Solutions: A Molecular Dynamics Study. Colloids Interfaces 2023, 7, 40. [Google Scholar] [CrossRef]
- Huang, C.; Quinn, D.; Sadovsky, Y.; Suresh, S.; Hsia, K.J. Formation and Size Distribution of Self-Assembled Vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, 2910–2915. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Discher, D.E.; Klein, M.L. Self-Assembly and Properties of Diblock Copolymers by Coarse-Grain Molecular Dynamics. Nat. Mater. 2004, 3, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Shelley, J.C.; Nielsen, S.O.; Discher, D.E.; Klein, M.L. Simulation of Diblock Copolymer Self-Assembly, Using a Coarse-Grain Model. J. Phys. Chem. B 2004, 108, 8153–8160. [Google Scholar] [CrossRef]
- Lipowsky, R. The Conformation of Membranes. Nature 1991, 349, 475–481. [Google Scholar] [CrossRef]
- Fromherz, P.; Röcker, C.; Rüppel, D. From discoid micelles to spherical vesicles. The concept of edge activity. Faraday Discuss. Chem. Soc. 1986, 81, 39–48. [Google Scholar] [CrossRef]
- Noguchi, H.; Gompper, G. Dynamics of vesicle self-assembly and dissolution. J. Chem. Phys. 2006, 125, 164908. [Google Scholar] [CrossRef]
- Yuan, H.; Huang, C.; Li, J.; Lykotrafitis, G.; Zhang, S. One-particle-thick, solvent-free, coarse-grained model for biological and biomimetic fluid membranes. Phys. Rev. E 2010, 82, 011905. [Google Scholar] [CrossRef]
- Canham, P.B. The Minimum Energy of Bending as a Possible Explanation of the Biconcave Shape of the Human Red Blood Cell. J. Theor. Biol. 1970, 26, 61–81. [Google Scholar] [CrossRef]
- Helfrich, W. Elastic Properties of Lipid Bilayers: Theory and Possible Experiments. Z. Naturforschung C 1973, 28, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Faucon, J.F.; Mitov, M.D.; Méléard, P.; Bivas, I.; Bothorel, P. Bending Elasticity and Thermal Fluctuations of Lipid Membranes-Theoretical and Experimental Requirements. J. Phys. 1989, 50, 2389–2414. [Google Scholar] [CrossRef]
- Evans, E.; Needham, D. Physical Properties of Surfactant Bilayer Membranes: Thermal Transitions, Elasticity, Rigidity, Cohesion and Colloidal Interactions. J. Phys. Chem. 1987, 91, 4219–4228. [Google Scholar] [CrossRef]
- Cooke, I.R.; Deserno, M. Coupling Between Lipid Shape and Membrane Curvature. Biophys. J. 2006, 91, 487–495. [Google Scholar] [CrossRef]
- Fogel, A.L.; Ravichandran, A.; Mani, S.; Upadhyay, B.; Khare, R.; Morgan, S.E. Water structure and mobility in acrylamide copolymer glycohydrogels with galactose and Siloxane Pendant Groups. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 584–597. [Google Scholar] [CrossRef]
- Walter, A.; Vinson, P.K.; Kaplun, A.; Talmon, Y. Intermediate structures in the cholate-phosphatidylcholine vesicle-micelle transition. Biophys. J. 1991, 60, 1315–1325. [Google Scholar] [CrossRef]
- Vinson, P.K.; Talmon, Y.; Walter, A. Vesicle-micelle transition of phosphatidylcholine and octyl glucoside elucidated by cryo-transmission electron microscopy. Biophys. J. 1989, 56, 669–681. [Google Scholar] [CrossRef]
- Davies, T.S.; Ketner, A.M.; Raghavan, S.R. Self-assembly of surfactant vesicles that transform into viscoelastic wormlike micelles upon heating. J. Am. Chem. Soc. 2006, 128, 6669–6675. [Google Scholar] [CrossRef]
- Markvoort, A.J.; van Santen, R.A.; Hilbers, P.A. Vesicle shapes from molecular dynamics simulations. J. Phys. Chem. B 2006, 110, 22780–22785. [Google Scholar] [CrossRef]
- Markvoort, A.J.; Pieterse, K.; Steijaert, M.N.; Spijker, P.; Hilbers, P.A. The Bilayer−Vesicle Transition is Entropy Driven. J. Phys. Chem. B 2005, 109, 22649–22654. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Schmid, F. Spontaneous formation of complex micelles from a homogeneous solution. Phys. Rev. Lett. 2008, 100, 137802. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Schmid, F. Dynamics of spontaneous vesicle formation in dilute solutions of Amphiphilic Diblock Copolymers. Macromolecules 2006, 39, 2654–2662. [Google Scholar] [CrossRef]
- Sevink, G.J.; Zvelindovsky, A.V. Self-assembly of complex vesicles. Macromolecules 2005, 38, 7502–7513. [Google Scholar] [CrossRef]
- Ye, X.; Khomami, B. Self-Assembly of Linear Diblock Copolymers in Selective Solvents: From Single Micelles to Particles with Tri-Continuous Inner Structures. Soft Matter 2020, 16, 6056–6062. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dormidontova, E.E. Equilibrium Chain Exchange Kinetics in Block Copolymer Micelle Solutions by Dissipative Particle Dynamics Simulations. Soft Matter 2011, 7, 4179. [Google Scholar] [CrossRef]
- Javan Nikkhah, S.; Turunen, E.; Lepo, A.; Ala-Nissila, T.; Sammalkorpi, M. Multicore assemblies from three-component linear homo-copolymer systems: A coarse-grained modeling study. Polymers 2021, 13, 2193. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, V.; Nielsen, S.O.; Discher, D.E.; Klein, M.L.; Lipowsky, R.; Shillcock, J. Dissipative particle dynamics simulations of polymersomes. J. Phys. Chem. B 2005, 109, 17708–17714. [Google Scholar] [CrossRef] [PubMed]
- Shillcock, J.C. Spontaneous vesicle self-assembly: A mesoscopic view of membrane dynamics. Langmuir 2012, 28, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, J.; Yang, J.; Wang, R.; Xie, D. Biomimetic membrane control of block copolymer vesicles with tunable wall thickness. Soft Matter 2013, 9, 2434. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Wang, Z.; Bao, M. Micelle-vesicle transitions in catanionic mixtures of SDS/DTAB induced by salt, temperature, and selective solvents: A dissipative particle dynamics simulation study. Colloid Polym. Sci. 2014, 292, 2349–2360. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Wang, B.; Jiang, J. Ph-sensitive vesicles formed by amphiphilic grafted copolymers with tunable membrane permeability for drug loading/release: A multiscale simulation study. Macromolecules 2016, 49, 6084–6094. [Google Scholar] [CrossRef]
- Wu, S.; Lu, T.; Guo, H. Dissipative particle dynamic simulation study of lipid membrane. Front. Chem. China 2010, 5, 288–298. [Google Scholar] [CrossRef]
- Feng, X.; Yan, N.; Jin, J.; Jiang, W. Disassembly of amphiphilic AB block copolymer vesicles in selective solvents: A molecular dynamics simulation study. Macromolecules 2023, 56, 2560–2567. [Google Scholar] [CrossRef]
- Lyubimov, I.; Wessels, M.G.; Jayaraman, A. Molecular dynamics simulation and prism theory study of assembly in solutions of amphiphilic bottlebrush block copolymers. Macromolecules 2018, 51, 7586–7599. [Google Scholar] [CrossRef]
- Chakraborty, K.; Shinoda, W.; Loverde, S.M. Molecular Simulation of the Shape Deformation of a Polymersome. Soft Matter 2020, 16, 3234–3244. [Google Scholar] [CrossRef]
- Sun, X.; Pei, S.; Wang, J.; Wang, P.; Liu, Z.; Zhang, J. Coarse-grained molecular dynamics simulation study on spherical and tube-like vesicles formed by amphiphilic copolymers. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 1220–1226. [Google Scholar] [CrossRef]
- Marrink, S.J.; Mark, A.E. Molecular dynamics simulation of the formation, structure, and dynamics of small phospholipid vesicles. J. Am. Chem. Soc. 2003, 125, 15233–15242. [Google Scholar] [CrossRef]
- Wu, R.; Deng, M.; Kong, B.; Yang, X. Coarse-grained molecular dynamics simulation of ammonium surfactant self-assemblies: Micelles and vesicles. J. Phys. Chem. B 2009, 113, 15010–15016. [Google Scholar] [CrossRef]
- Sambasivam, A.; Dhakal, S.; Sureshkumar, R. Structure and Rheology of Self-Assembled Aqueous Suspensions of Nanoparticles and Wormlike Micelles. Mol. Simul. 2017, 44, 485–493. [Google Scholar] [CrossRef]
- Sambasivam, A.; Sangwai, A.V.; Sureshkumar, R. Self-Assembly of Nanoparticle–Surfactant Complexes with Rodlike Micelles: A Molecular Dynamics Study. Langmuir 2016, 32, 1214–1219. [Google Scholar] [CrossRef]
- Sambasivam, A.; Sangwai, A.V.; Sureshkumar, R. Dynamics and Scission of Rodlike Cationic Surfactant Micelles in Shear Flow. Phys. Rev. Lett. 2015, 114, 8302. [Google Scholar] [CrossRef]
- Dhakal, S.; Sureshkumar, R. Anomalous Diffusion and Stress Relaxation in Surfactant Micelles. Phys. Rev. E 2017, 96, 2605. [Google Scholar] [CrossRef]
- Dhakal, S.; Sureshkumar, R. Uniaxial Extension of Surfactant Micelles: Counterion Mediated Chain Stiffening and a Mechanism of Rupture by Flow-Induced Energy Redistribution. ACS Macro Lett. 2015, 5, 108–111. [Google Scholar] [CrossRef]
- Dhakal, S.; Sureshkumar, R. Topology, Length Scales, and Energetics of Surfactant Micelles. J. Chem. Phys. 2015, 143, 024905. [Google Scholar] [CrossRef] [PubMed]
- Sangwai, A.V.; Sureshkumar, R. Binary Interactions and Salt-Induced Coalescence of Spherical Micelles of Cationic Surfactants From Molecular Dynamics Simulations. Langmuir 2011, 28, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Sangwai, A.V.; Sureshkumar, R. Coarse-Grained Molecular Dynamics Simulations of the Sphere to Rod Transition in Surfactant Micelles. Langmuir 2011, 27, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The Martini Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Darré, L.; Machado, M.R.; Pantano, S. Coarse-grained models of water. WIREs Comput. Mol. Sci. 2012, 2, 921–930. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to Dot surface contouring of Molecular Assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]
- Pratt, V. Direct least-squares fitting of algebraic surfaces. ACM SIGGRAPH Comput. Graph. 1987, 21, 145–152. [Google Scholar] [CrossRef]
- Park, S.Y.; Bera, A.K. Maximum Entropy Autoregressive conditional heteroskedasticity model. J. Econom. 2009, 150, 219–230. [Google Scholar] [CrossRef]
- Yang, Y. Structure, Dynamics and Rheology of Polymer Solutions from Coarse-Grained Molecular Dynamics: Effects of Polymer Concentration, Solvent Quality and Geometric Confinement. Ph.D. Dissertation, Syracuse University, Syracuse, NY, USA, 2015. [Google Scholar]
- Lee, H.; de Vries, A.H.; Marrink, S.-J.; Pastor, R.W. A coarse-grained model for polyethylene oxide and polyethylene glycol: Conformation and hydrodynamics. J. Phys. Chem. B 2009, 113, 13186–13194. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.P.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to An External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Pair Correlation Function | Probability Distribution Function, PEO | Probability Distribution Function, PB |
|---|---|---|---|
Wormlike micelle |  |  |  |
Rectangular lamella |  |  |  |
Disk lamella |  |  |  |
Cavity micelle |  |  |  |
Vesicle |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Sureshkumar, R. Energetic and Entropic Motifs in Vesicle Morphogenesis in Amphiphilic Diblock Copolymer Solutions. Colloids Interfaces 2024, 8, 12. https://doi.org/10.3390/colloids8010012
Liu S, Sureshkumar R. Energetic and Entropic Motifs in Vesicle Morphogenesis in Amphiphilic Diblock Copolymer Solutions. Colloids and Interfaces. 2024; 8(1):12. https://doi.org/10.3390/colloids8010012
Chicago/Turabian StyleLiu, Senyuan, and Radhakrishna Sureshkumar. 2024. "Energetic and Entropic Motifs in Vesicle Morphogenesis in Amphiphilic Diblock Copolymer Solutions" Colloids and Interfaces 8, no. 1: 12. https://doi.org/10.3390/colloids8010012
APA StyleLiu, S., & Sureshkumar, R. (2024). Energetic and Entropic Motifs in Vesicle Morphogenesis in Amphiphilic Diblock Copolymer Solutions. Colloids and Interfaces, 8(1), 12. https://doi.org/10.3390/colloids8010012