1. Introduction
Layered calcium phosphates (octacalcium phosphate Ca
8(HPO
4)
4(PO
4)
2·5H
2O (OCP), brushite CaHPO
4·2H
2O) are calcium hydroxyapatite Ca
10(PO
4)
6(OH)
2 (HAp) crystallization precursors, and therefore attract attention as promising materials for bone tissue replacement and regeneration due to the higher rate of dissolution in the body (resorption) compared to HAP-based materials [
1].
In their structure, calcium alkyl phosphates are rather similar to OCP [
2], i.e., consisting of alternating hydrated and alkyl-containing layers. Due to the layered structure, their particles have a lamellar morphology, which leads to high values of the specific surface area and sorption capacity, as well as to potentially more efficient reinforcement of polymer matrices compared to equiaxed particles. Nucleic acids, phospholipids, and other organic phosphates play an important role in the life of the body. Alkyl phosphates form cell walls, such as the vesicular membrane of phospholipids, and play an important role as sources of hard tissues.
In the last 50 years, alkyl phosphate derivatives, such as dialkyl phosphates, monoalkylphosphatidylethanol amines, and monoalkylphosphocholines, have become important compounds used in food chemistry, medicinal chemistry, supramolecular chemistry, and geochemistry [
3]. An interesting aspect of the use of monoalkyl phosphates is the surface modification of inorganic materials, such as HAp [
4,
5] or titanium implants [
6]. It was shown that an increase in the chain length (from propyl phosphate to hexadecyl phosphate) promotes better crystallization of HAP on the modified surface of a titanium implant due to a higher density of Ca
2+-binding sites stem from carboxylic acids. In addition, it should be noted that alkyl phosphates, including branched ones, are used as surfactants [
7] as well as self-assembled monolayers [
8].
Among the methods for the synthesis of alkyl phosphates with an alkyl chain length from C
4 to C
12, the following can be distinguished: the interaction of pyrophosphoric acid and the corresponding alcohol in benzene [
9]; the interaction of alcohol with phosphorus oxychloride in the presence of dimethylaniline and subsequent interaction with sodium methylate [
10]; interaction of phosphorus oxide P
2O
5 with alcohol [
11], in which case a mixture of mono- and dialkyl phosphate is obtained; interaction of acetyl phosphate obtained from a mixture of phosphoric acid with acetic anhydride in the presence of pyridine and triethanolamine, with alcohol [
12]; interaction of alcohol with diphenyl phosphorochloridate, etc. [
3].
In addition to using layered phosphates themselves as individual biomaterials [
13], or functional components [
14] in composites, they are promising as precursors to make biphasic ceramics based on calcium pyrophosphate Ca
2P
2O
7 (CPP) and tricalcium phosphate Ca
3(PO
4)
2 (TCP) [
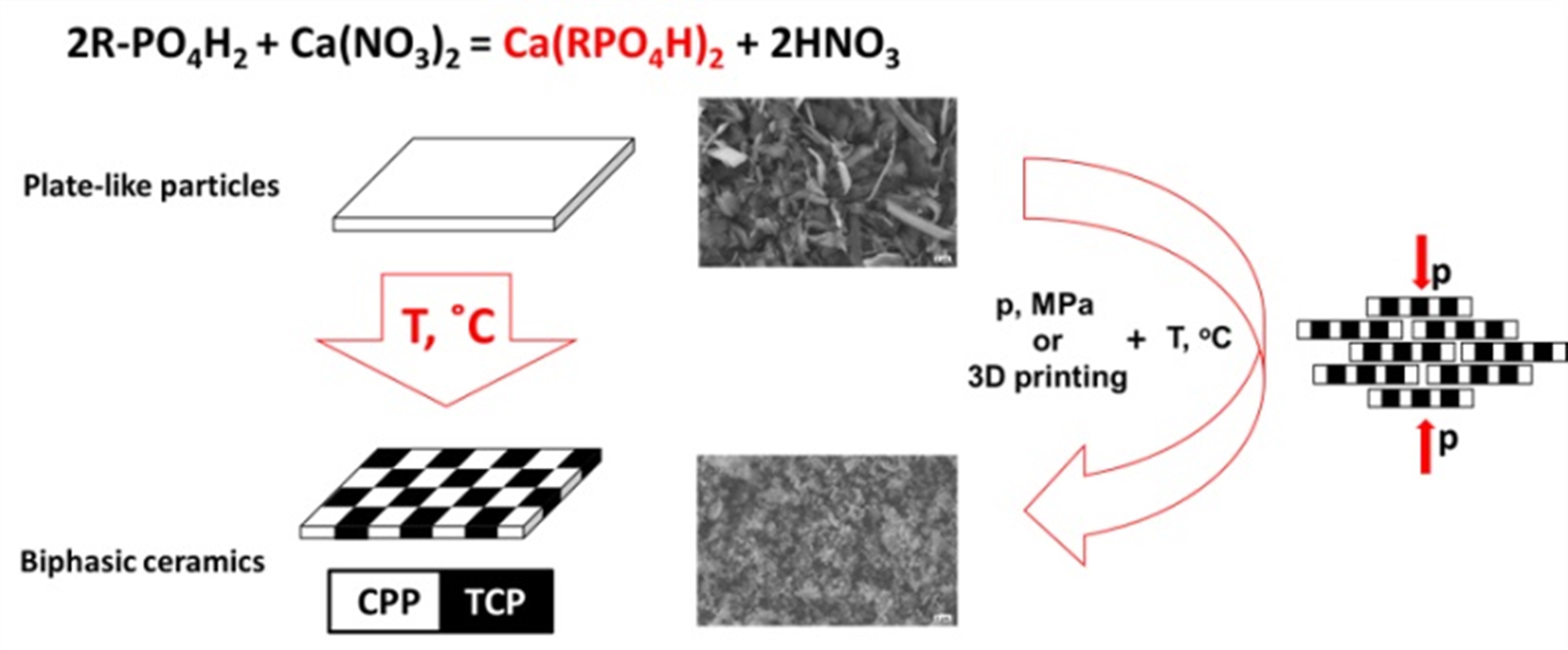
15] with homogeneous distribution of the two phases. The lamellar morphology of the alkyl phosphate particles leads to the formation of a structure similar to a “brickwall” (in comparison with to spherical particles or particles of arbitrary shape) and contributes to the better moldability of their powders, leading to dense, tough bioceramics after firing. The molding of biomaterials based on layered phosphates could be realized not only by traditional methods, such as isostatic pressing, but additive manufacturing techniques, e.g., ink-jet printing [
16] and stereolithography [
14], in which case they affect the optical properties and viscosity of the initial mixtures and mechanical and functional properties of the resulting material.
In this work, calcium alkyl phosphates are considered as analogues of layered phosphates which could be used as precursors of calcium phosphate ceramics with Ca/P < 1.5. Considering the thermal behavior of calcium alkyl phosphates with alkyl chain lengths n = 4, 8 and 12 (butyl, octyl, and dodecyl, respectively), the structure and micromorphology of the obtained powders and thermolyzed products were studied.
2. Materials and Methods
The following reagents were used during the synthesis: butanol-1 (SigmaAldrich, Munchen, Germany, 99.9%), octanol-1 (SigmaAldrich, Munchen, Germany, for synthesis), dodecanol-1 (SigmaAldrich, Munchen, Germany, for synthesis), benzene (SigmaAldrich, ≥99.0%), pyrophosphoric acid (synthesized or SigmaAldrich, Munchen, Germany, tech.), diethyl ether (analytical grade), sodium hydroxide (Labtech, analytical grade), hydrochloric acid (SigmaTec, Moscow, Russia, high purity grade), distilled water. The synthesis of alkyl phosphoric acids was carried out as described in [
9]. Briefly, alcohols were added to the obtained pyrophosphoric acid in an equimolar ratio, or a small excess of acid was used. Since dodecyl alcohol has a near-room melting point (24–27 °C), during the synthesis it is necessary to use an inert solvent C
6H
6 benzene, while for liquid alcohols (butanol-1 and octanol-1) its use is optional. All preparations were carried out at room temperature, because higher temperatures can result in a small degree of dehydration of the alcohol to an alkene and melting of the pyrophosphoric acid. The reaction took place at room temperature under constant stirring for at least 48 hours. Since it was not possible to wait for the completion of the reaction, which is probably of second order, it was necessary to separate the product from the unreacted alcohol and pyrophosphoric acid, in addition to the co-product orthophosphoric acid. After dissolving the reaction solution in ether and one or two aqueous extractions to remove inorganic acids, the monoalkyl phosphate was extracted into an aqueous solution of sodium hydroxide NaOH at pH 12 by transformation into a water-soluble monoalkyl disodium phosphate, which allowed the unreacted alcohol to be recovered from its ether solution. The pH of the aqueous medium was then adjusted to 1 with concentrated hydrochloric acid. At pH 1, the monoalkyl phosphoric acid was recovered by extraction into ether. The ether was evaporated in air for 1 day, after which the product was additionally dried in a vacuum desiccator for 30 minutes. Reactions proceeded according to the equation:
where R = butyl (But), octyl (Oct), dodecyl (Dod).
The synthesis of calcium alkyl phosphates was carried out by precipitation from aqueous solutions containing alkyl phosphoric acids and calcium salt. The calcium salt was 4-aqueous calcium nitrate Ca(NO3)2·4H2O (SigmaAldrich, Munchen, Germany, ≥99.0%).
During the synthesis of calcium alkyl phosphates, an aqueous solution of calcium nitrate was added to an aqueous solution of alkyl phosphate. In addition to the synthesized alkyl phosphates, commercial dibutyl phosphate (SigmaAldrich, Munchen, Germany, 97%) was used as a reagent. The mixture was stirred for 2 hours at room temperature. After stirring, the solution was evaporated in an oven at 100 °C for 24 h. The reaction proceeded according to the following scheme:
The heat treatment of obtained calcium alkyl phosphates was carried out in alumina crucibles at muffle furnace Nabertherm L 9/12 (Nabertherm GmbH, Lilienthal, Germany) up to 400, 600, and 1000 °C with a heating rate of 5 °C/min and 1 h dwelling time.
Thermogravimetric (TG) and differential thermal (DTA) analysis was carried out on a STA 409 PC Luxx thermal analyzer (Netzsch, Selb, Germany). The heating rate was 5 °C/min, temperature range 40–1400 °C, in an air atmosphere. The composition of the exhaust gases was studied using an Aëolos QMS 403 C quadrupole mass spectrometer (Netzsch, Selb, Germany) with heating of the inlet capillary system up to 300 °C. The sample for analysis was about 10 mg in mass and placed in alundum crucibles.
X-ray studies were carried out on a Rigaku D/Max-2500 diffractometer with a rotating anode. The spectra were recorded in reflection mode in standard Bragg-Brentano geometry using CuKα (λ = 1.54183 Å). The following XRD-spectra were chosen to acquire: angle 2θ range = 2–60°, 2θ step = 0.02°, spectra recording rate = 5°/min. Qualitative analysis of the obtained radiographs was carried out using the WinXPOW program using cards from the ICDD PDF-2 database.
The as-synthesized and heat-treated powders were studied using a scanning electron microscope LEO SUPRA 50VP with a field emission source (Carl Zeiss, Oberkochen, Germany). The samples were placed onto a copper plate using a conductive carbon adhesive with a chromium layer of 10–15 nm deposited (Quorum Technologies sputtering unit, QT-150T ES, East Sussex, UK). The accelerating voltage of the electron gun was 5–21 kV. Images were obtained in secondary electrons at magnifications up to 50,000 using a detector type SE2. Electron probe microanalysis was carried out on a LEO SUPRA 50VP microscope (Carl Zeiss, Oberkochen, Germany) equipped with an INCA Energy 300 energy dispersive system-ray spectrometer (EDX) (Oxford Instruments, Abingdon, Great Britain). The analysis was carried out with an accelerating voltage of 20 kV. For each sample, the obtained data were averaged at least for 10 points and over the entire image area.
The IR absorption spectra of the samples were recorded on a Spectrum One IR spectrophotometer (Perkin Elmer, Waltham, MA, USA) in the range 550–4000 cm
–1 with a scanning step of 1 cm
–1 in the attenuated total reflection mode. The spectra were analyzed based on published data [
17]. The spectra were processed using the Spectrum software.
3. Results and Discussion
Among the criteria to choose a method for the synthesis of alkyl phosphates the following ones were considered: (i) the availability and cheapness of reagents while maintaining high purity; (ii) the possibility of synthesis under simple conditions (without the use of an inert medium or high temperatures); (iii) the number of references to the technique in the literature. From this viewpoint, we have adopted the method of synthesis from pyrophosphoric acid and alcohol in a benzene medium. In the case of using butanol-1, a yellowish liquid product was obtained; interaction with octanol-1 gave a slightly yellow gel-like product; and a white solid product was obtained in the case of dodecanol-1.
The targeting butyl, dibutyl, octyl, and dodecyl calcium phosphate represent white powders. XRD was used to obtain diffraction patterns of the synthesized compounds, which could not be identified from existing crystallographic databases (
Figure 1). The positions of the XRD reflections in this work differ from the literature data, which confirms the lack of experimental data on such compounds. It should be noted that the presence of reflections in the low-angle 2θ region (from 2 to 6°) confirms the layered structure of the alkyl phosphates under study resembling the X-ray diffraction pattern of OCP (ICDD PDF-2 card number [74-1301]; 2θ (100) = 4.701°, d(100) = 18.781 Å).The structure of thecobtained powders probably is similar to the structure of octacalcium phosphate where the apatite and brushite layers alternate each other [
2]. In calcium alkyl phosphates, the apatite layer alternates with alkyl layer, in which alkyl tails could be oriented by a random angle to the (h00) plane. The interplanar spacing corresponding to the first three reflections in the diffraction patterns of the obtained calcium alkyl phosphates linearly depends on the number of carbon atoms in the alkyl chain, and correlates as 3 to 2 to 1 (Figure 3), which indicates that the crystallographic planes are parallel and, apparently, correspond to reflexes (h00), where h = 1, 2, 3 (or 2, 4, 6, etc.). Partial data on the diffraction patterns of calcium octyl and dodecyl phosphates can be found in articles [
4,
18], with calcium oxide and calcium chloride as source of Ca
2+-cations, respectively.
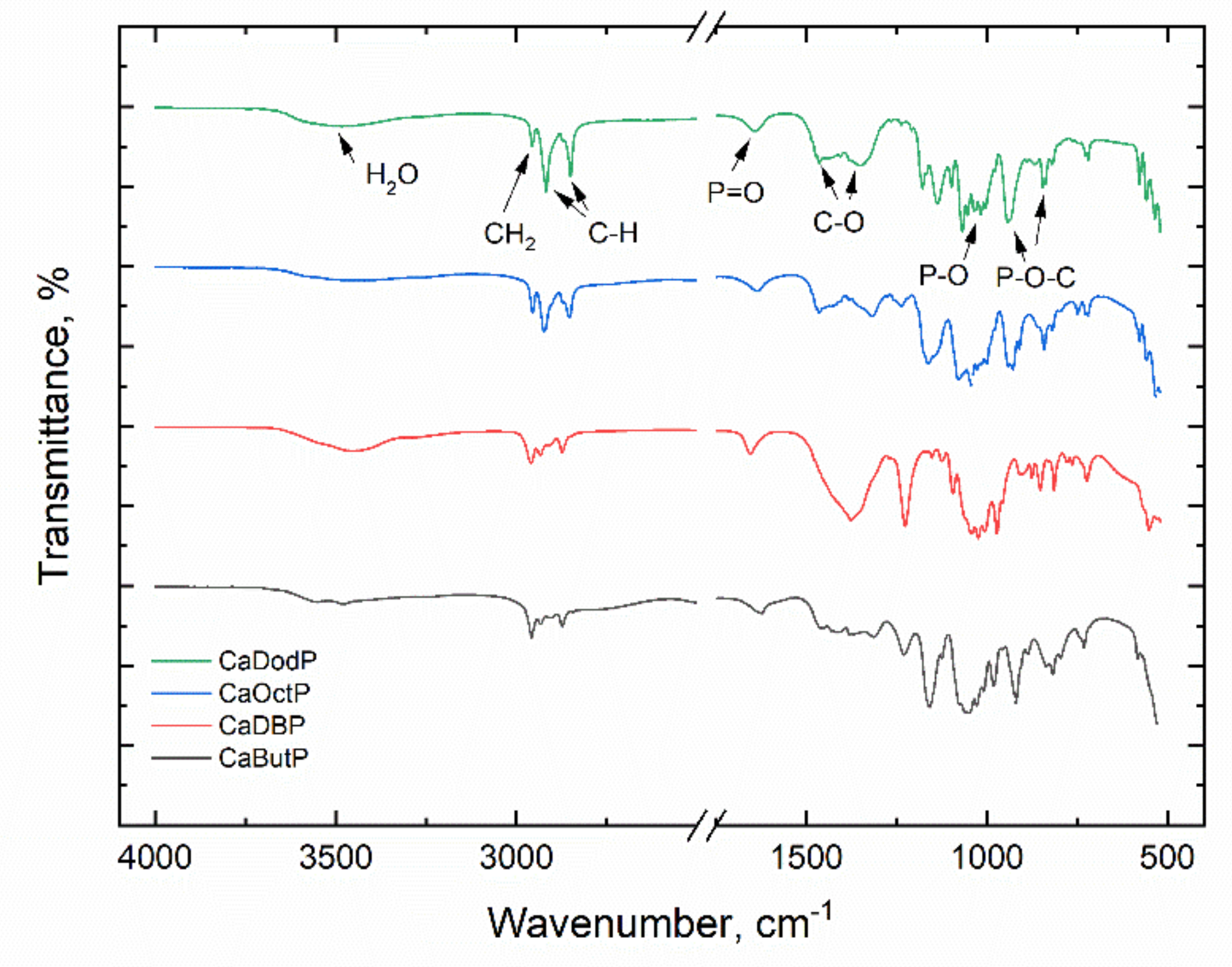
IR spectra of the obtained calcium alkyl phosphates (
Figure 2) show a wide absorption band at 3460 cm
−1 which related to the vibration of crystallization water. Three bands in the range of 2850–2960 cm
−1 corresponds to the stretching vibrations of C-H groups. Two bands (or one wide band in case of calcium dibutyl phosphate (CaDBP)) in the range of 1410–1450 cm
−1 could be associated with vibrations of C-O group, and a lot of bands below 1200 cm
−1 relate to stretching and bending vibrations of P-O, P=O, and P-O-C groups.
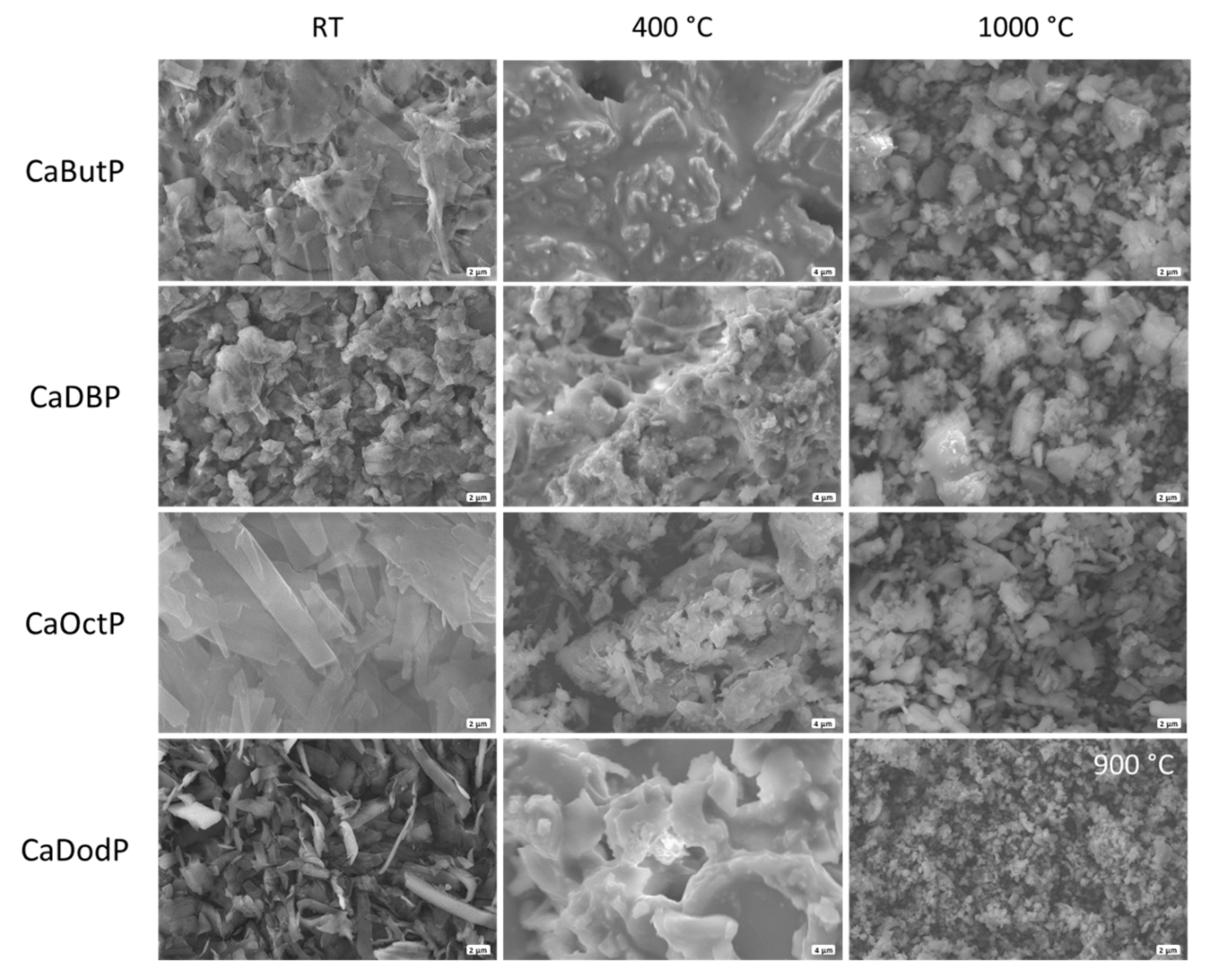
By means of scanning electron microscopy, it was found that the powders of all calcium alkyl phosphates have a plate-like morphology (
Figure 3) similar to the morphology of layered OCP. At the same time, butyl and dodecyl calcium phosphate powders are prone to agglomeration. To the touch, the powders of the obtained alkyl phosphates and their calcium salts are soap-like, approving their use as surface-active substances. According to the data of EDX analysis (
Table 1), the atomic ratios of Ca to P (close to 1–2) were established, which apparently indicates the formation of crystalline hydrates (due to an excess of oxygen atoms) of acid salts of calcium alkyl phosphates. Formation of acid salts could be related to the accumulation of nitric acid during the syntheses (2–3) as well as with the initially low pH values of these solutions.
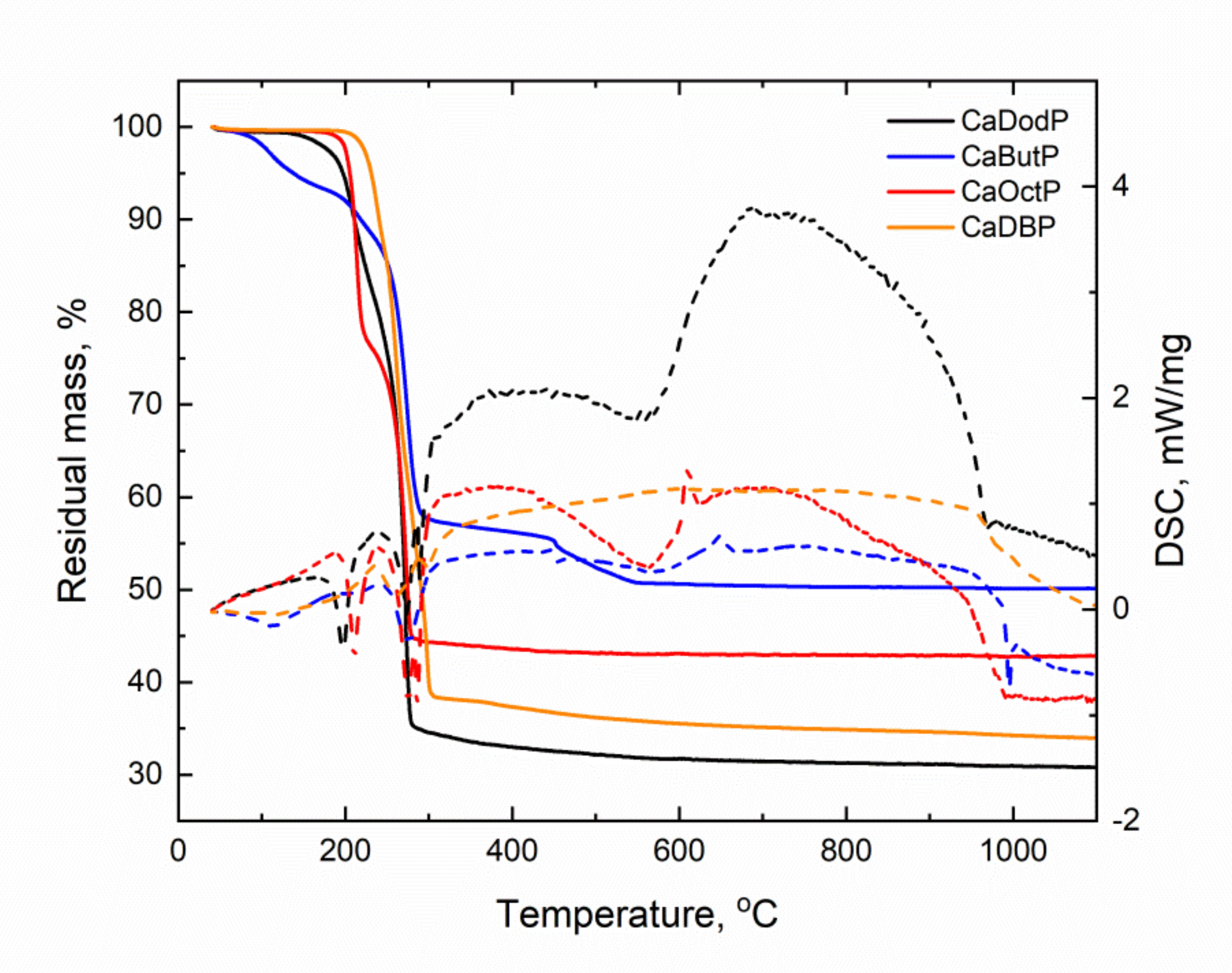
Simultaneous thermal analysis showed that decomposition occurs in two stages for calcium dibutyl, octyl, and dodecyl phosphate, and in three stages for butyl phosphate (
Figure 4). All materials decompose at up to 278–280 °C (octyl, dodecyl), 290 °C (butyl), and 300 °C (dibutyl), associated with the cleavage of the alkyl chain, combustion (and, perhaps, pyrolysis) of the resulting chain fragment and the release of carbon dioxide and water vapor. In this case, the dark color of the powders is preserved up to 600 °C. The values of residual masses do not allow to determine the type of calcium phosphate forming because, for both assumed calcium metaphosphate (for acid salts) and calcium pyrophosphate (for base salts), the experimental mass loss values exceed the theoretical ones (
Table 2). Mass spectrometry of released gases shows the removal of water and carbon dioxide at each stage of decomposition. The DSC curves have two narrow endothermic peaks at 190–205 °C and 270–290 °C, and two broad exothermic peaks in range of 300–550 °C and 600–970 °C.
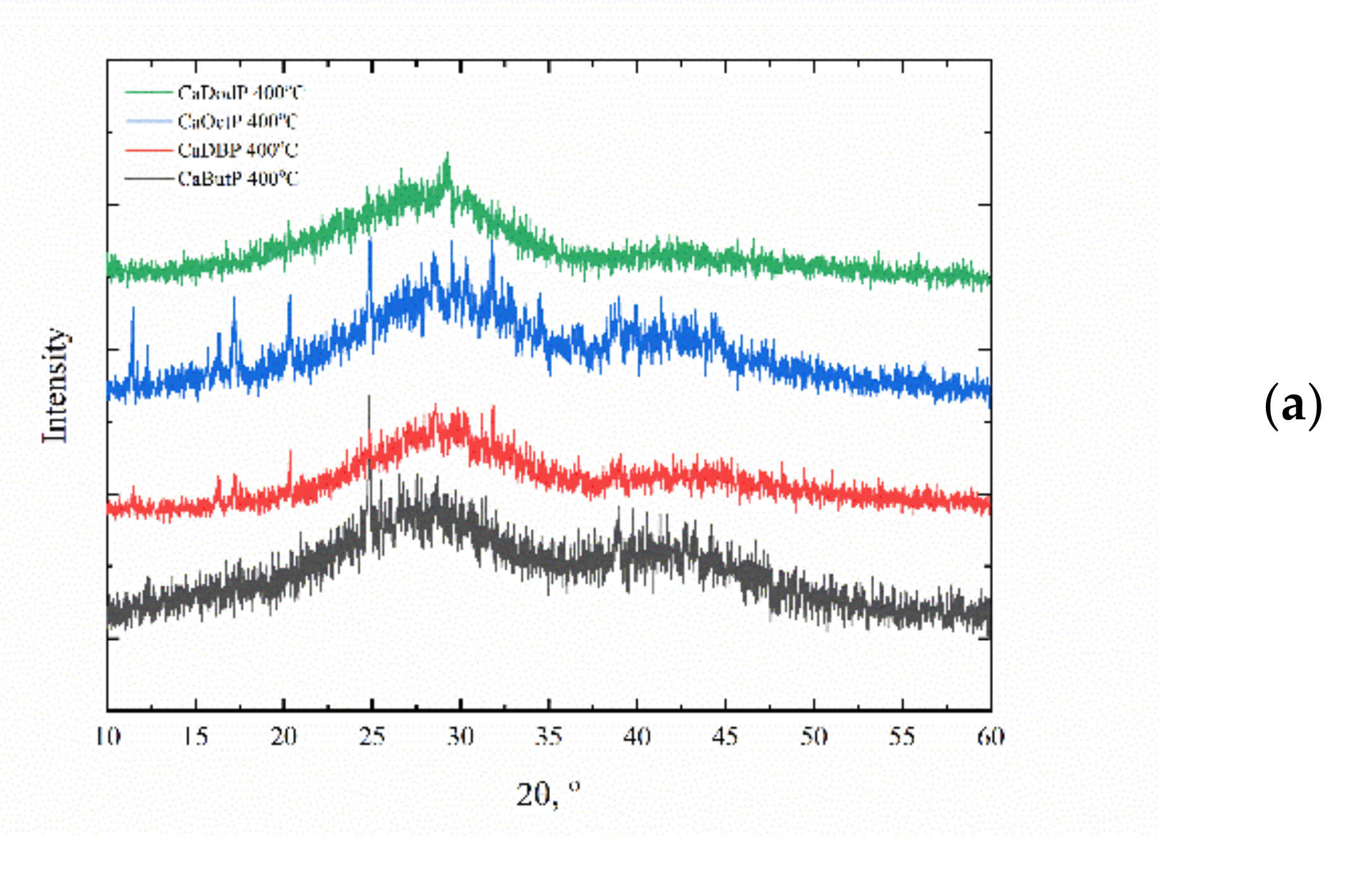
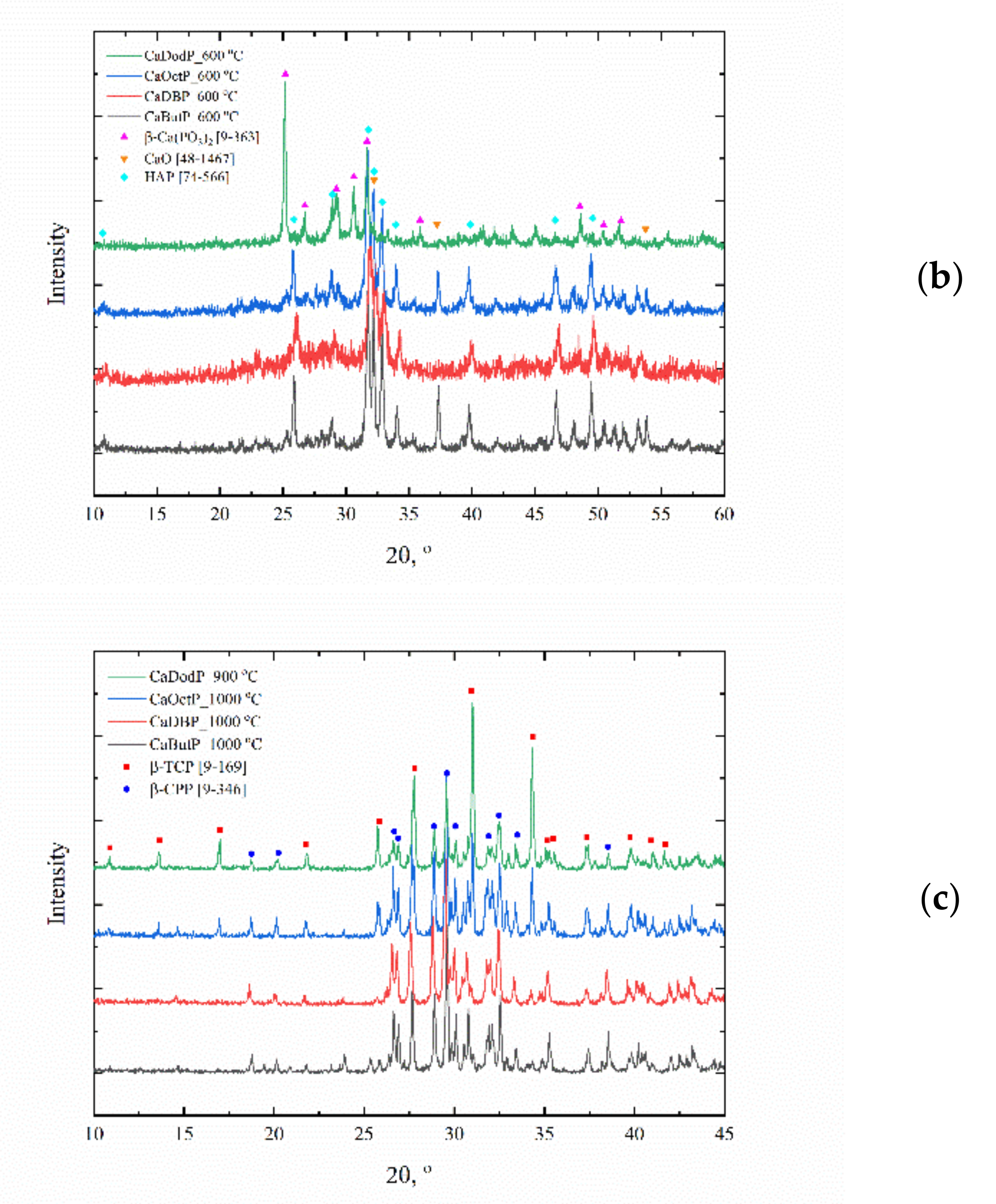
For clarifying the composition of obtained calcium alkyl phosphates, the phase composition of thermolyzed products was investigated. After 400 °C (
Figure 5a), the X-ray amorphous products were obtained with a halo at 2θ = 25–30 ° coming from vitreous calcium phosphate and residual carbon. The morphology of thermolyzed powders inherited from initial ones (
Figure 6) without any evidence of water evaporation and carbon dioxide release from its composition, except calcium butyl phosphate, for which the plate-like particles were destroyed and presumably homogeneously mixed with formed carbon. The presence of residual carbon and respective dark color of such powders could be used for the formulation of the material composition for the stereolithography 3D printing of calcium phosphate filled materials where such powders can act both as functional filler and as photoabsorbent additive (dye) to compensate light scattering. At 600 °C (
Figure 5b), the formation of the mixture of HAp (Ca/P = 1.67) and calcium metaphosphate Ca(PO
3)
2 (Ca/P = 0.5) with a small quantity of calcium oxide was occurred. Moreover, at 1000 °C (
Figure 5c), this mixture converts into the mixture of β-TCP (Ca/P = 1.5) and β-CPP (Ca/P = 1). The morphology of powders at 1000 °C changes to randomly shaped particles with submicron size. Calcium dodecyl phosphate which melts at 1000 °C in corundum crucible (and at 969 °C during STA—
Figure 4), was fired at 900 °C and has the smallest particle size (about 300–400 nm) which could be perspective regarding fabrication of dense bioceramics. The phase evolution of resulting calcium phosphates, especially after 400 °C, when the mass of all studied powders is almost constant according to TG, could be explained by pyrohydrolysis of the phosphates and probable release of phosphoric acid vapours from alkyl phosphates at high temperatures or solid-state reaction of HAp and calcium metaphosphate accompanied by the exothermic effects observed in
Figure 4 (on DSC curves).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}