
Indigo Carmine Degradation in Water Induced by a Pulsed Positive Corona Discharge in Air: Discharge and Postdischarge Effects

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pulsed Positive Corona Discharge
2.2. Treatments and Statistical Analysis
2.3. Physicochemical Determinations
2.3.1. IC Degradation
2.3.2. pH and Electrical Conductivity
2.3.3. Hydrogen Peroxide Measurement
2.3.4. Nitrate Measurement
2.3.5. Nitrite Measurement
2.4. Modeling of IC Degradation in the Postdischarge Phase
3. Results and Discussion
3.1. pH and Electrical Conductivity in PAW
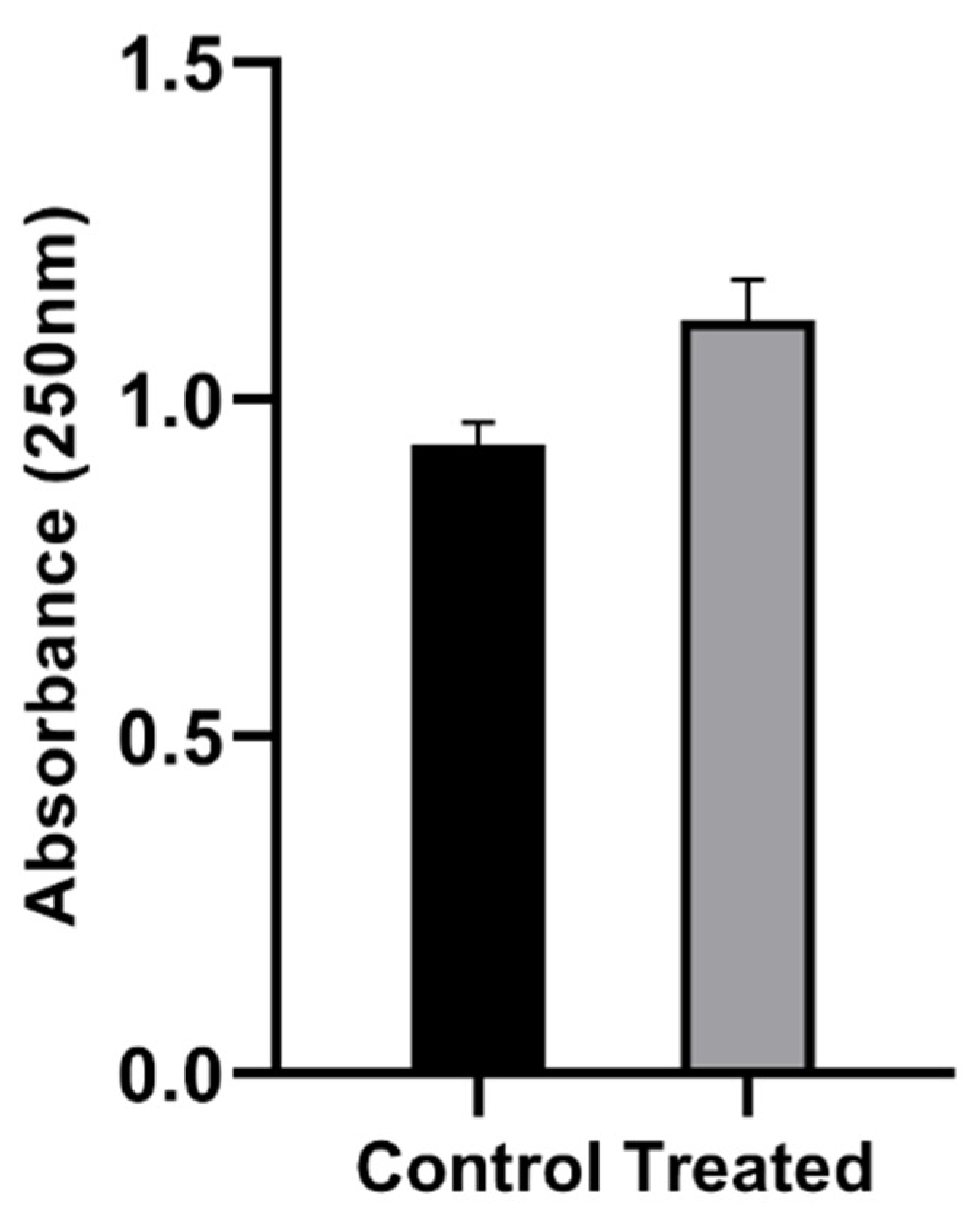
3.2. Concentration of Reactive Species in PAW
3.2.1. Hydrogen Peroxide
3.2.2. Nitrate and Nitrite
3.3. Time Evolution of the Concentration of Reactive Species in the Postdischarge
3.4. IC Degradation: Discharge and Postdischarge Effects
3.4.1. Modeling of the IC Degradation in the Postdischarge Phase
3.4.2. Subproducts of the IC Degradation
3.5. Energy Yield G50
4. Conclusions
- The OH˙ radical plays a primary role in the IC degradation. While both O3 and OH˙ contribute to the IC degradation in the discharge phase, the OH˙ radical contributes in the postdischarge phase. While both O3 and OH˙ are mainly generated in the gas phase and then transferred to the liquid in the discharge phase, the OH˙ radical is mainly produced through the O=NOOH decomposition in the postdischarge phase. Noticeably, increases in the energy yield values are observed at 24 h post-treatment due to the beneficial effects induced by the O=NOOH decomposition.
- Maximum values for the degradation of the IC unsaturated bond at 0 h were obtained for a plasma exposure time of 30 min: 81.3% and 86% for sample volumes of 100 mL and 50 mL, respectively; while corresponding values for the IC chromogenic bond at the same plasma exposure time were 93.4% and 99.1% for sample volumes of 100 mL and 50 mL, respectively.
- For low plasma exposure times (<20 min) the IC degradation of both the chromogenic and unsaturated bonds at 24 h post-treatment was higher than 0 h; being the IC degradation for 50 mL higher than 100 mL. However, for higher plasma exposure times the IC degradation was almost independent of both the volume treated and the post-treatment time.
- Concentrations of reactive species in the aqueous phase at 0 h increased with the plasma exposure time and decreased with increasing sample volume. The maximum values reached being 7.25 mg/L for H2O2, 15 mg/L for NO3− and 2 mg/L for NO2−. While the concentrations of NO2− and H2O2 decreased with post-treatment time, the NO3− concentration remained stable.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, F.F. Erratum to: Introduction to Plasma Physics and Controlled Fusion, 3rd ed.; Springer International Publishing: Los Angeles, CA, USA, 2018; ISBN 9783319223087. [Google Scholar]
- Raizer, Y.P. Gas Discharge Physics, 1st ed.; Allen, J.E., Ed.; Springer: Berlin/Heidelberg, Germany, 1991; ISBN 978-3-642-64760-4. [Google Scholar]
- Boulos, M.I.; Fauchais, P.; Pfender, E. Thermal Plasmas: Fundamentals and Applications, 1st ed.; Springer: New York, NY, USA, 1994; ISBN 9781489913395. [Google Scholar]
- Fridman, A.; Chirokov, A.; Gutsol, A. Non-thermal atmospheric pressure discharges. J. Phys. D Appl. Phys. 2005, 38, R1–R24. [Google Scholar] [CrossRef]
- Adamovich, I.; Baalrud, S.D.; Bogaerts, A.; Bruggeman, P.J.; Cappelli, M.; Colombo, V.; Czarnetzki, U.; Ebert, U.; Eden, J.G.; Favia, P.; et al. The 2017 Plasma Roadmap: Low temperature plasma science and technology. J. Phys. D Appl. Phys. 2017, 50, 323001. [Google Scholar] [CrossRef]
- Wright, K. Study of Plasma Treatment of Produced Water from Oil and Gas Exploration; Drexel University: Philadelphia, PA, USA, 2015. [Google Scholar]
- Cho, Y.I.; Wright, K.C.; Kim, H.S.; Cho, D.J.; Rabinovich, A.; Fridman, A. Stretched arc discharge in produced water. Rev. Sci. Instrum. 2015, 86, 013501. [Google Scholar] [CrossRef] [PubMed]
- Manakhov, A.; Orlov, M.; Grokhovsky, V.; Alghunaimi, F.I.; Ayirala, S. Functionalized Nanomembranes and Plasma Technologies for Produced Water Treatment: A Review. Polymers 2022, 14, 1785. [Google Scholar] [CrossRef]
- Choi, S.; Watanabe, T. Decomposition of 1-decanol emulsion by water thermal plasma jet. IEEE Trans. Plasma Sci. 2012, 40, 2831–2836. [Google Scholar] [CrossRef]
- Crema, A.P.S.; Piazza Borges, L.D.; Micke, G.A.; Debacher, N.A. Degradation of indigo carmine in water induced by non-thermal plasma, ozone and hydrogen peroxide: A comparative study and by-product identification. Chemosphere 2020, 244, 125502. [Google Scholar] [CrossRef]
- Wright, K. Plasma Water Treatment and Oxidation of Organic Matter in Water. In Proceedings of the IEEE International Pulsed Power Conference, Orlando, FL, USA, 23–29 June 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 35–38. [Google Scholar]
- Machala, Z.; Tarabová, B.; Sersenová, D.; Janda, M.; Hensel, K. Chemical and antibacterial effects of plasma activated water: Correlation with gaseous and aqueous reactive oxygen and nitrogen species, plasma sources and air flow conditions. J. Phys. D Appl. Phys. 2019, 52, 034002. [Google Scholar] [CrossRef]
- Anderson, C.E.; Cha, N.R.; Lindsay, A.D.; Clark, D.S.; Graves, D.B. The Role of Interfacial Reactions in Determining Plasma–Liquid Chemistry. Plasma Chem. Plasma Process 2016, 36, 1393–1415. [Google Scholar] [CrossRef]
- Foster, J.E. Plasma-based water purification: Challenges and prospects for the future. Phys. Plasmas 2017, 24, 055501. [Google Scholar] [CrossRef]
- Bruggeman, P.J.; Kushner, M.J.; Locke, B.R.; Gardeniers, J.G.E.; Graham, W.G.; Graves, D.B.; Hofman-Caris, R.C.H.M.; Maric, D.; Reid, J.P.; Ceriani, E.; et al. Plasma-liquid interactions: A review and roadmap. Plasma Sources Sci. Technol. 2016, 25, 053002. [Google Scholar] [CrossRef]
- Malik, M.A. Water purification by plasmas: Which reactors are most energy efficient? Plasma Chem. Plasma Process 2010, 30, 21–31. [Google Scholar] [CrossRef]
- Lukes, P.; Locke, B.R.; Brisset, J.L. Aqueous-Phase Chemistry of Electrical Discharge Plasma in Water and in Gas-Liquid Environments. In Plasma Chemistry and Catalysis in Gases and Liquids; Parvulescu, V.I., Magureanu, M., Lukes, P., Eds.; 2012; pp. 243–308. ISBN 9783527330065. [Google Scholar]
- Kumar, A.; Škoro, N.; Gernjak, W.; Puač, N. Cold atmospheric plasma technology for removal of organic micropollutants from wastewater—A review. Eur. Phys. J. D 2021, 75, 283. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Jin, G.; Li, Y.; Fangchuan, Z.; Pingdao, G. Degradation of dye wastewater by ns-Pulse dbd plasma. Plasma Sci. Technol. 2013, 15, 928–934. [Google Scholar] [CrossRef]
- Baird, R.B.; Eaton, A.D.; Rice, E.W. Standard Methods for the Examination of Water and Wastewater, 23rd ed.; Baird, R.B., Eaton, A.D., Rice, E.W., Eds.; American Water Works Association; American Public Works Association; Water Environment Federation: Washington, DC, USA, 2017; ISBN 9780123821652. [Google Scholar]
- Lukes, P.; Dolezalova, E.; Sisrova, I.; Clupek, M. Aqueous-phase chemistry and bactericidal effects from an air discharge plasma in contact with water: Evidence for the formation of peroxynitrite through a pseudo-second-order post-discharge reaction of H2O2 and HNO2. Plasma Sources Sci. Technol. 2014, 23, 015019. [Google Scholar] [CrossRef]
- Julák, J.; Scholtz, V.; Kotúčová, S.; Janoušková, O. The persistent microbicidal effect in water exposed to the corona discharge. Phys. Med. 2012, 28, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Vlad, I.E.; Anghel, S.D. Time stability of water activated by different on-liquid atmospheric pressure plasmas. J. Electrostat. 2017, 87, 284–292. [Google Scholar] [CrossRef]
- Zhao, Y.M.; Ojha, S.; Burgess, C.M.; Sun, D.W.; Tiwari, B.K. Inactivation efficacy and mechanisms of plasma activated water on bacteria in planktonic state. J. Appl. Microbiol. 2020, 129, 1248–1260. [Google Scholar] [CrossRef]
- Traylor, M.J.; Pavlovich, M.J.; Karim, S.; Hait, P.; Sakiyama, Y.; Clark, D.S.; Graves, D.B. Long-term antibacterial efficacy of air plasma-activated water. J. Phys. D Appl. Phys. 2011, 44, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Minamitani, Y.; Shoji, S.; Ohba, Y.; Higashiyama, Y. Decomposition of dye in water solution by pulsed power discharge in a water droplet spray. IEEE Trans. Plasma Sci. 2008, 36, 2586–2591. [Google Scholar] [CrossRef]
- Damschen, D.E.; Martin, L.R. Aqueous aerosol oxidation of nitrous acid by O2, O3 AND H2O2. Atmos. Environ. 1983, 17, 2005–2011. [Google Scholar] [CrossRef]
- Saha, A.; Goldstein, S.; Cabelli, D.; Czapski, G. Determination of optimal conditions for synthesis of peroxynitrite by mixing acidified hydrogen peroxide with nitrite. Free Radic. Biol. Med. 1998, 24, 653–659. [Google Scholar] [CrossRef]
- Schönekerl, S.; Weigert, A.; Uhlig, S.; Wellner, K.; Pörschke, R.; Pfefferkorn, C.; Backhaus, K.; Lerch, A. Evaluating the performance of a lab-scale water treatment plant using non-thermal plasma technology. Water 2020, 12, 1956. [Google Scholar] [CrossRef]
- Cano, M.; Solis, M.; Diaz, J.; Solis, A.; Loera, O.; Teutli, M.M. Biotransformation of indigo carmine to isatin sulfonic acid by lyophilized mycelia from trametes versicolor. Afr. J. Biotechnol. 2011, 10, 12224–12231. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Post-Treatment Time | G50 [g/kWh] | |||
|---|---|---|---|---|
| (at 285 nm) | (at 610 nm) | |||
| 50 mL | 100 mL | 50 mL | 100 mL | |
| 0 h | 5.2 | 5.2 | 6 | 6.2 |
| 24 h | 15.5 | 8.5 | 17 | 9.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreyra, M.G.; Fina, B.L.; Milardovich, N.J.; Chamorro, J.C.; Santamaría, B.; Balestrasse, K.; Prevosto, L. Indigo Carmine Degradation in Water Induced by a Pulsed Positive Corona Discharge in Air: Discharge and Postdischarge Effects. Plasma 2022, 5, 265-279. https://doi.org/10.3390/plasma5020021
Ferreyra MG, Fina BL, Milardovich NJ, Chamorro JC, Santamaría B, Balestrasse K, Prevosto L. Indigo Carmine Degradation in Water Induced by a Pulsed Positive Corona Discharge in Air: Discharge and Postdischarge Effects. Plasma. 2022; 5(2):265-279. https://doi.org/10.3390/plasma5020021
Chicago/Turabian StyleFerreyra, Matías G., Brenda L. Fina, Natalio J. Milardovich, Juan C. Chamorro, Brenda Santamaría, Karina Balestrasse, and Leandro Prevosto. 2022. "Indigo Carmine Degradation in Water Induced by a Pulsed Positive Corona Discharge in Air: Discharge and Postdischarge Effects" Plasma 5, no. 2: 265-279. https://doi.org/10.3390/plasma5020021
APA StyleFerreyra, M. G., Fina, B. L., Milardovich, N. J., Chamorro, J. C., Santamaría, B., Balestrasse, K., & Prevosto, L. (2022). Indigo Carmine Degradation in Water Induced by a Pulsed Positive Corona Discharge in Air: Discharge and Postdischarge Effects. Plasma, 5(2), 265-279. https://doi.org/10.3390/plasma5020021