
N-4 Alkyl Cytosine Derivatives Synthesis: A New Approach

 , , ,
, , , 
 , and
, and 
Abstract
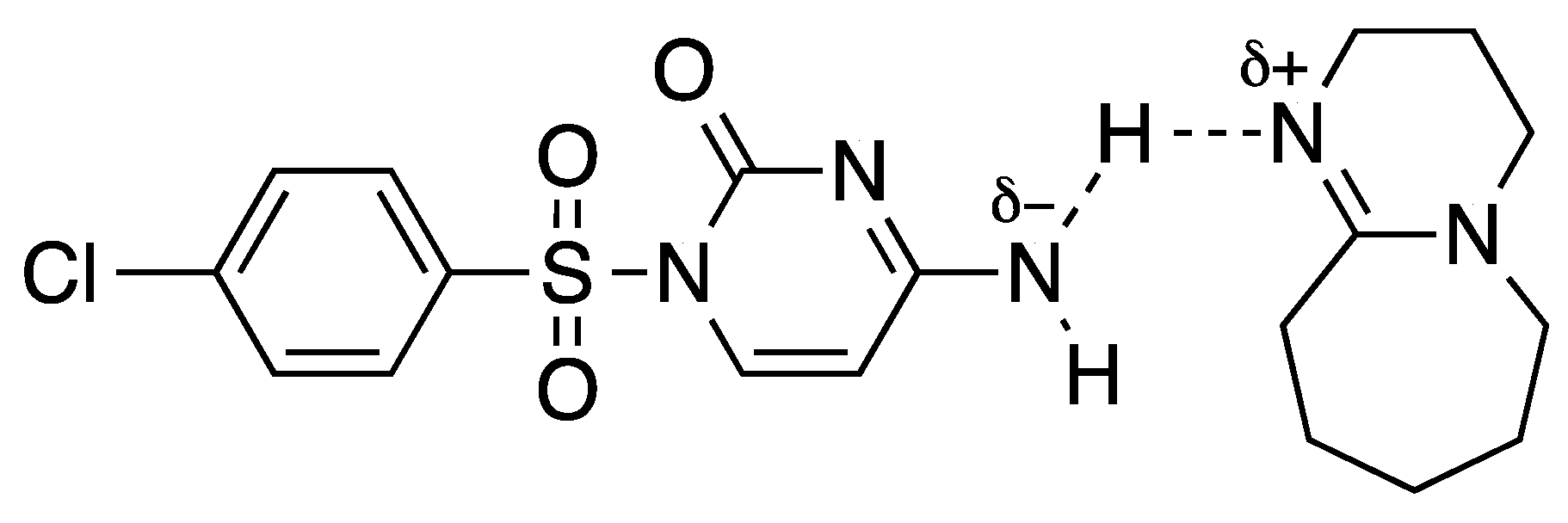
:1. Introduction
2. Materials and Methods
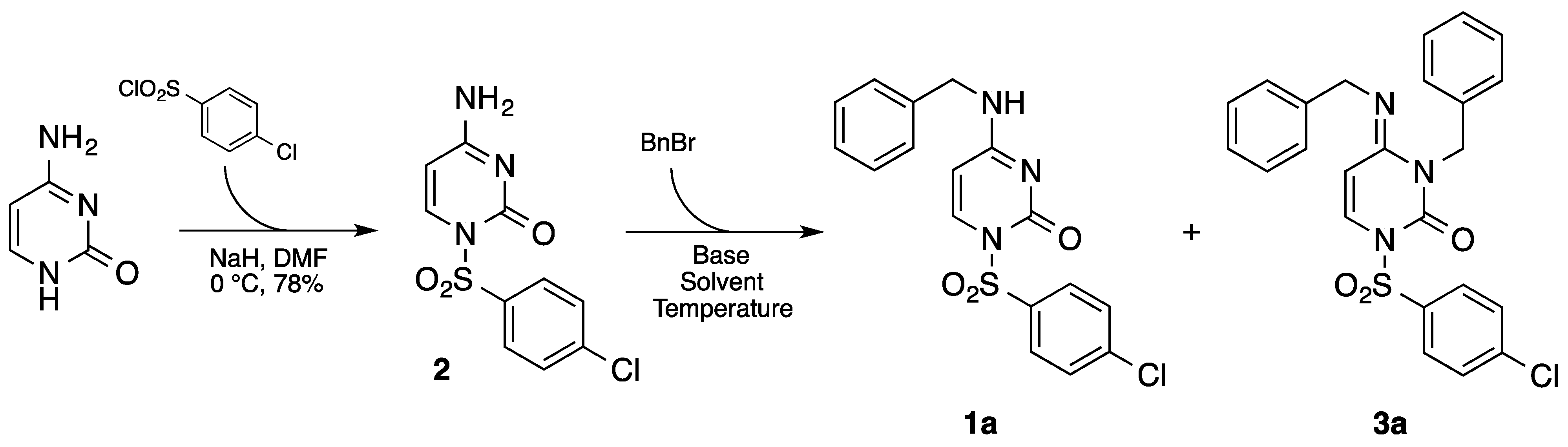
2.1. Synthesis of 4-amino-1-((4-chlorophenyl)sulfonyl) pyrimidin-2(1H)-one (2)
2.2. General Procedure Synthesis of N-4 Alkyl Cytosine Derivatives
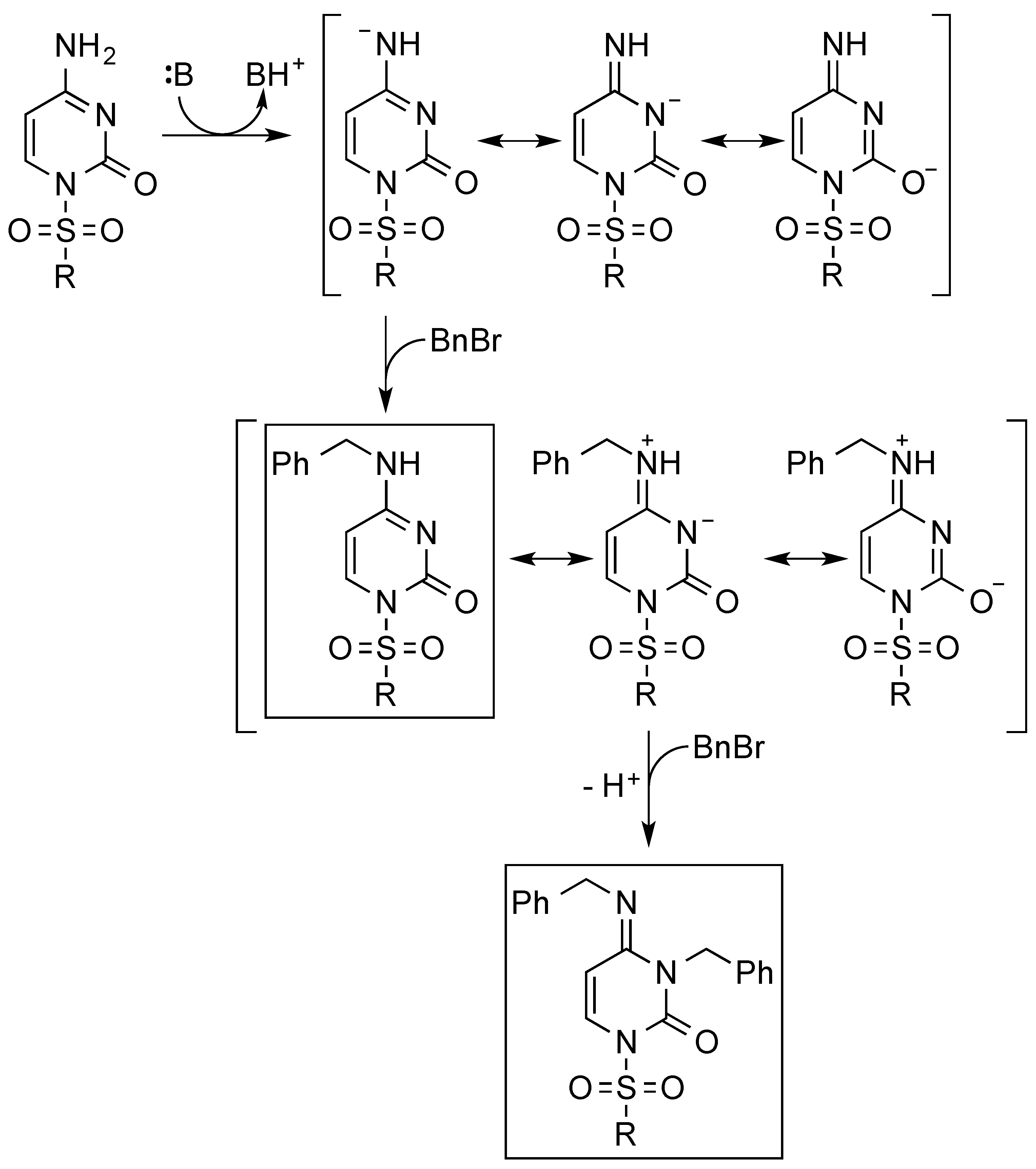
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Espeseth, A.S.; Felock, P.; Wolfe, A.; Witmer, M.; Grobler, J.; Anthony, N.; Egbertson, M.; Melamed, J.Y.; Young, S.; Hamill, T.; et al. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. USA 2000, 97, 11244–11249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Dobard, C.M.; Chow, S.A. Requirement for Integrase during Reverse Transcription of Human Immunodeficiency Virus Type 1 and the Effect of Cysteine Mutations of Integrase on its Interactions with Reverse Transcriptase. J. Virol. 2004, 78, 5045–5055. [Google Scholar] [CrossRef] [Green Version]
- McColl, D.J.; Chen, X. Strand transfer inhibitors of HIV-1 integrase: Bringing IN a new era of antiretroviral therapy. Antivir. Res. 2010, 85, 101–118. [Google Scholar] [CrossRef]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef] [PubMed]
- Anker, M.; Corales, R.B. Raltegravir (MK-0518): A novel integrase inhibitor for the treatment of HIV infection. Expert Opin. Investig. Drugs 2008, 17, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sax, P.E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J.E.; Liu, H.C.; Zhong, L.; Yale, K.; et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 2012, 379, 2439–2448. [Google Scholar] [CrossRef]
- Shimura, K.; Kodama, E.; Sakagami, Y.; Matsuzaki, Y.; Watanabe, W.; Yamataka, K.; Watanabe, Y.; Ohata, Y.; Doi, S.; Sato, M.; et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Motomura, T.; Aramaki, H.; Matsuda, T.; Yamashita, M.; Ito, Y.; Kawakami, H.; Matsuzaki, Y.; Watanabe, W.; Yamataka, K.; et al. Novel HIV-1 Integrase Inhibitors Derived from Quinolone Antibiotics. J. Med. Chem. 2006, 49, 1506–1508. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef]
- Cuzzucoli Crucitti, G.; Pescatori, L.; Messore, A.; Madia, V.M.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Di Leva, F.S.; Cosconati, S.; et al. Discovery of N-aryl-naphthylamines as in vitro inhibitors of the interaction between HIV integrase and the cofactor LEDGF/p75. Eur. J. Med. Chem. 2015, 101, 288–294. [Google Scholar] [CrossRef]
- Engelman, A.; Kessl, J.J.; Kvaratskhelia, M. Allosteric inhibition of HIV-1 integrase activity. Curr. Opin. Chem. Biol. 2013, 17, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, L.; Ferro, S.; Morreale, F.; De Grazia, A.; Chimirri, S. Inhibitors of the Interactions between HIV-1 IN and the Cofactor LEDGF/p75. ChemMedChem 2011, 6, 1184–1191. [Google Scholar] [CrossRef]
- Hare, S.; Vos, A.M.; Clayton, R.F.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20057–20062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasuji, T.; Johns, B.A.; Yoshida, H.; Taishi, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.; Yoshinaga, T.; Seki, T.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors. 1. Molecular design and establishment of an advanced two-metal binding pharmacophore. J. Med. Chem. 2012, 55, 8735–8744. [Google Scholar] [CrossRef]
- Pendri, A.; Meanwell, N.A.; Peese, K.M.; Walker, M.A. New first and second generation inhibitors of human immunodeficiency virus-1 integrase. Expert Opin. Ther. Pat. 2011, 21, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Johns, B.A.; Svolto, A.C. Advances in two-metal chelation inhibitors of HIV integrase. Expert Opin. Ther. Pat. 2008, 18, 1225–1237. [Google Scholar] [CrossRef]
- Di Santo, R. Inhibiting the HIV Integration Process: Past, Present, and the Future. J. Med. Chem. 2014, 57, 539–566. [Google Scholar] [CrossRef]
- Baumann, R.; Baxendale, I.R. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintori, C. University of Siena, Siena, Italy. Unpublished work, 2022.
- Cesar Gonzalez, C.; de Cabrera, M.; Wnuk, S.F. Gemcitabine analogues with 4-N-alkyl chain modified with fluoromethyl ketone group. Nucleosides Nucleotides Nucleic Acids 2018, 37, 248–260. [Google Scholar] [CrossRef]
- Quercia, R.; Perno, C.F.; Koteff, J.; Moore, K.; McCoig, C.; St. Clair, M.; Kuritzkes, D.J. Twenty-Five Years of Lamivudine: Current and Future Use for the Treatment of HIV-1 Infection. Acquir. Immune Defic. Syndr. 2018, 78, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Keane, S.J.; Ford, A.; Mullins, N.D.; Maguire, N.M.; Legigan, T.; Balzarini, J.; Maguire, A.R. Design and Synthesis of α-Carboxy Nucleoside Phosphonate Analogues and Evaluation as HIV-1 Reverse Transcriptase-Targeting Agents. J. Org. Chem. 2015, 80, 2479–2493. [Google Scholar] [CrossRef]
- Masaki; K.; Takayuki, S.; Isamu, K. Bicyclic cytosine derivative-containing double strand nucleic acids interfering HIV replication. JP 2012179027 A, 20 September 2012.
- Hossain, N.; Rozenski, J.; Clercq, E.D.; Herdewijn, P. Synthesis and Antiviral Activity of the α-Analogues of 1,5-Anhydrohexitol Nucleosides (1,5-Anhydro-2,3-dideoxy-d-ribohexitol Nucleosides). J. Org. Chem. 1997, 62, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Tanaka, M.; Fukushima, M.; Sasaki, T.; Matsuda, A. Nucleosides and Nucleotides. 158. 1-(3-C-Ethynyl-β-d-ribo-pentofuranosyl)-cytosine, 1-(3-C-Ethynyl-β-d-ribopentofuranosyl)uracil, and Their Nucleobase Analogues as New Potential Multifunctional Antitumor Nucleosides with a Broad Spectrum of Activity. J. Med. Chem. 1996, 39, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Badawey, E.; Kappe, T. Synthesis of some new imidazo[1,2-a]pyrimidin-5(1H)-ones as potential antineoplastic agents. J. Heterocycl. Chem. 1995, 32, 1003–1006. [Google Scholar] [CrossRef]
- Joseph, H.; Burke, J.M. Optimization of an Anti-HIV Hairpin Ribozyme by in Vitro Selection. J. Biol. Chem. 1993, 268, 24515–24518. [Google Scholar] [CrossRef]
- Güzel-Akdemir, Ö.; Akdemir, A.; Pan, P.; Vermelho, A.B.; Parkkila, S.; Scozzafava, A.; Capasso, C.; Supuran, C.T. A Class of Sulfonamides with Strong Inhibitory Action against the α-Carbonic Anhydrase from Trypanosoma cruzi. J. Med. Chem. 2013, 56, 5773–5781. [Google Scholar] [CrossRef] [PubMed]
- Tayebee, R.; Nehzat, F. A Simple and Effective Methodology for the Sulfonylation of Alcohols and Aniline under Solvent Free Condition at Room Temperature. Am. J. Med. Sci. 2012, 2, 36–39. [Google Scholar] [CrossRef] [Green Version]
- Barcelò-Oliver, M.; Baquero, B.A.; Bauzà, A.; Garcìa-Raso, A.; Terròn, A.; Mata, I.; Molinsb, E.; Frontera, A. Experimental and theoretical study of thymine and cytosine derivatives: The crucial role of weak noncovalent interactions. CrystEngComm 2012, 14, 5777–5784. [Google Scholar] [CrossRef]
- Kašnar-Šamprec, J.; Ratkaj, I.; Mišković, K.; Pavlak, M.; Baus-Lončar, M.; Kraljević Pavelić, S.; Glavaš-Obrovac, L.; Žinić, B. In vivo toxicity study of N-1-sulfonylcytosine derivatives and their mechanisms of action in cervical carcinoma cell line. Investig. New Drugs 2012, 30, 981–990. [Google Scholar] [CrossRef]
- Spijker, H.J.; van Delft, F.L.; van Hest, J.C.M. Atom Transfer Radical Polymerization of Adenine, Thymine, Cytosine, and Guanine Nucleobase Monomers. Macromolecules 2007, 40, 12–18. [Google Scholar] [CrossRef]
- Glavaš-Obrovac, L.; Karner, I.; Štefanic, M.; Kašnar-Šamprec, J.; Žinic, B. Metabolic effects of novel N-1-sulfonylpyrimidine derivatives on human colon carcinoma cells. Il Farmaco 2005, 60, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Plitta, B.; Adamska, E.; Giel-Pietraszuk, M.; Fedoruk-Wyszomirska, A.; Naskret-Barciszewska, M.; Markiewicz, W.T.; Barciszewski, J. New cytosine derivatives as inhibitors of DNA methylation. Eur. J. Med. Chem. 2012, 55, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Mattson, R.J.; Pham, K.M.; Leuck, D.J.; Cower, K.A. An improved method for reductive alkylation of amines using titanium(IV) isopropoxide and sodium cyanoborohydride. J. Org. Chem. 1990, 55, 2552–2554. [Google Scholar] [CrossRef]
- Borch, R.F. Reductive Amination with sodium cyanoborohydride: N,N-dimethylcyclohexylamine. Org. Synth. 1972, 52, 124–125. [Google Scholar]
- Borch, R.F.; Hassid, A.I. New method for the methylation of amines. J. Org. Chem. 1972, 37, 1673–1674. [Google Scholar] [CrossRef]
- Borch, R.F.; Bernstein, M.D.; Durst, H.D. Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Shapiro, R.; Weisgras, J.M. Bisulfite-catalyzed transamination of cytosine and cytidine. Biochem. Biophys. Res. Commun. 1970, 40, 839–843. [Google Scholar] [CrossRef]
- Kasnar-Samprec, J.; Glavas-Obrovac, L.; Pavlak, M.; Mihaljevic, I.; Mrljak, V.; Stambuk, N.; Konjevoda, P.; Zinic, B. Synthesis, Spectroscopic Characterization and Biological Activity of N-1-Sulfonylcytosine Derivatives. Croat. Chem. Acta 2005, 78, 261–267. [Google Scholar]
- Sun, T.; Darbre, F.; Keese, R. Synthesis of vitamin B12 derivatives incorporating peripheral cytosine and N-acetylcytosine. Tetrahedron 1999, 55, 9777–9786. [Google Scholar] [CrossRef]
- Kašnar, B.; Krizmanić, I.; Žinića, M. Synthesis of the Sulfonylpyrimidine Derivatives as a New Type of Sulfonylcycloureas. Nucleosides Nucleotides 1997, 16, 1067–1071. [Google Scholar] [CrossRef]
- Nand, B.; Khanna, G.; Chaudhary, A.; Lumb, A.; Khurana, J.M. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU): A Versatile Reagent in Organic Synthesis. Curr. Org. Chem. 2015, 19, 790–812. [Google Scholar] [CrossRef]
- Jagrut, L.B.; Waghmare, R.A.; Mane, R.A.; Jadhav, W.N. An Improved Synthetic Route for the Synthesis of Sulfonamides. Int. J. ChemTech Res. 2011, 3, 1592–1595. [Google Scholar]
- Timmerman, J.C.; Widenhoefer, R.A. Gold-Catalyzed Intermolecular anti-Markovnikov Hydroamination of Methylenecyclopropanes with 2-Pyridones. Adv. Synth. Catal. 2015, 357, 3703–3706. [Google Scholar] [CrossRef]
- Tsuyoshi, M.; Gunzi, S.; Kazukuni, N.; Yuichiro, E.; Genki, H.; Yasuhito, S.; Shogo, M.; Masafumi, S.; Drozdova, O.O.; Kyuya, Y. Complex Formation between a Nucleobase and Tetracyanoquinodimethane Derivatives: Crystal Structures and Transport Properties of Charge-Transfer Solids of Cytosine. Bull. Chem. Soc. Jpn. 2008, 81, 331–344. [Google Scholar]
- Freccero, M.; Di Valentin, C.; Sarzi-Amade, M. Modeling H-Bonding and Solvent Effects in the Alkylation of Pyrimidine Bases by a Prototype Quinone Methide: A DFT Study. J. Am. Chem. Soc. 2003, 125, 3544–3553. [Google Scholar] [CrossRef]
- Kawabata, T.; Majumdar, S.; Tsubaki, D.; Monguchi, D. Memory of chirality in intramolecular conjugate addition of enolates: A novel access to nitrogen heterocycles with contiguous quaternary and tertiary stereocenters. Org. Biol. Chem. 2005, 3, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.Y.; Moschel, R.C. Effect of Ionic State of 2’-Deoxyguanosine and Solvent on Its Aralkylation by Benzyl Bromide. Chem. Res. Toxicol. 1998, 11, 696–702. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Base 1 | Solvent | T (°C) | 1a (%) | 3a (%) | Time (h) 2 |
|---|---|---|---|---|---|---|
| 1 | DMSO | 25→80 | ND | ND | 24 | |
| 2 | Pyr | DMSO | 25→80 | <1 | ND | 24 |
| 3 | Pyr | DMF | 25→80 | <1 | ND | 24 |
| 4 | TEA | DMF | 25→80 | <1 | ND | 24 |
| 5 | DBU | DMF | 25 | <1 | Trace | 24 |
| 6 | LDA | DMF | −40 | 5 | 3 | 24 |
| 7 | LiHMDS | DMF | −40 | 3.5 | 2.5 | 4 |
| 8 | KHMDS | DMF | 0 | 16 | 9 | 4 |
| 9 | KHMDS | DMF | −23 | 30 | 18 | 4 |
| 10 | KHMDS | DMF | −40 | 40 | 30 | 4 |
| 11 | KHMDS | DMF | −60 | 20 | 15 | 4 |
| Entry | Electrophile 1 | Product 1 (%) | Product 3 (%) | Ratio 1:3 |
|---|---|---|---|---|
| b |  | 41 | 26 | 6:4 |
| c |  | 32 | 20 | 6:4 |
| d |  | 32 | 25 | 6:4 |
| e |  | 23 | 8 | 7:3 |
| f |  | 32 | 21 | 6:4 |
| g |  | 31 | 19 | 6:4 |
| h |  | 13 | 5 | 7:3 |
| i | CH3Br | 20 | ND | --- |
| Entry | Base 1 | Solvent (1:1) | T (°C) | 1a (%) | 3a (%) | Time (h) |
|---|---|---|---|---|---|---|
| 1 | KHMDS | DMF/THF | −40 | 39 | 26 | 4 |
| 2 | KHMDS | CH2Cl2/THF | −40 | 4 | 1 | 4 |
| 3 | KHMDS | CH2Cl2/THF | −40→5 | 9 | 2 | 4 |
| 4 | KHMDS | CH2Cl2/THF | −40→5 | 77 | 17 | 24 |
| Entry | Electrophile 1 | Product 1 (%) | Product 3 (%) | Ratio 1:3 |
|---|---|---|---|---|
| 1 |  | 57 | 14 | 8:2 |
| 2 |  | 47 | 5 | 9:1 |
| 3 |  | 40 | 4 | 9:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Nisco, M.; Di Maio, A.; Manfra, M.; Ostacolo, C.; Bertamino, A.; Campiglia, P.; Gomez-Monterrey, I.M.; Pedatella, S. N-4 Alkyl Cytosine Derivatives Synthesis: A New Approach. Reactions 2022, 3, 192-202. https://doi.org/10.3390/reactions3010014
De Nisco M, Di Maio A, Manfra M, Ostacolo C, Bertamino A, Campiglia P, Gomez-Monterrey IM, Pedatella S. N-4 Alkyl Cytosine Derivatives Synthesis: A New Approach. Reactions. 2022; 3(1):192-202. https://doi.org/10.3390/reactions3010014
Chicago/Turabian StyleDe Nisco, Mauro, Antonio Di Maio, Michele Manfra, Carmine Ostacolo, Alessia Bertamino, Pietro Campiglia, Isabel M. Gomez-Monterrey, and Silvana Pedatella. 2022. "N-4 Alkyl Cytosine Derivatives Synthesis: A New Approach" Reactions 3, no. 1: 192-202. https://doi.org/10.3390/reactions3010014
APA StyleDe Nisco, M., Di Maio, A., Manfra, M., Ostacolo, C., Bertamino, A., Campiglia, P., Gomez-Monterrey, I. M., & Pedatella, S. (2022). N-4 Alkyl Cytosine Derivatives Synthesis: A New Approach. Reactions, 3(1), 192-202. https://doi.org/10.3390/reactions3010014