
High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts


Abstract
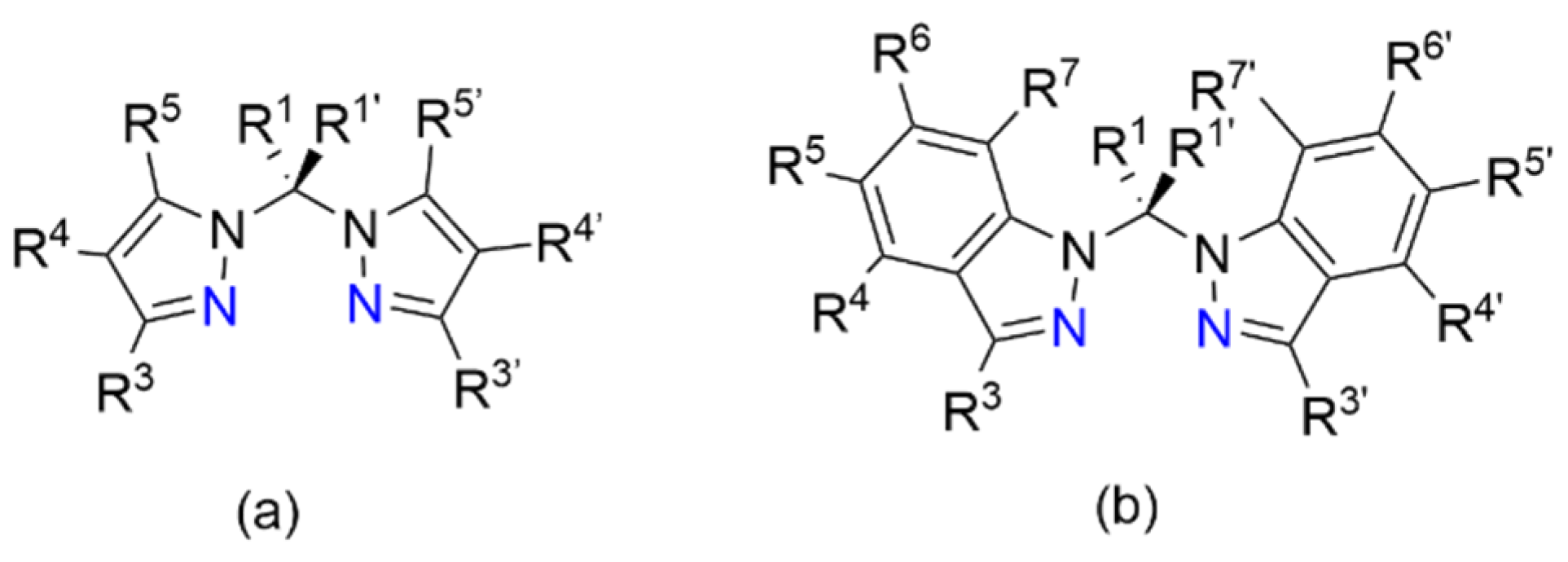
:1. Introduction
2. Materials and Methods
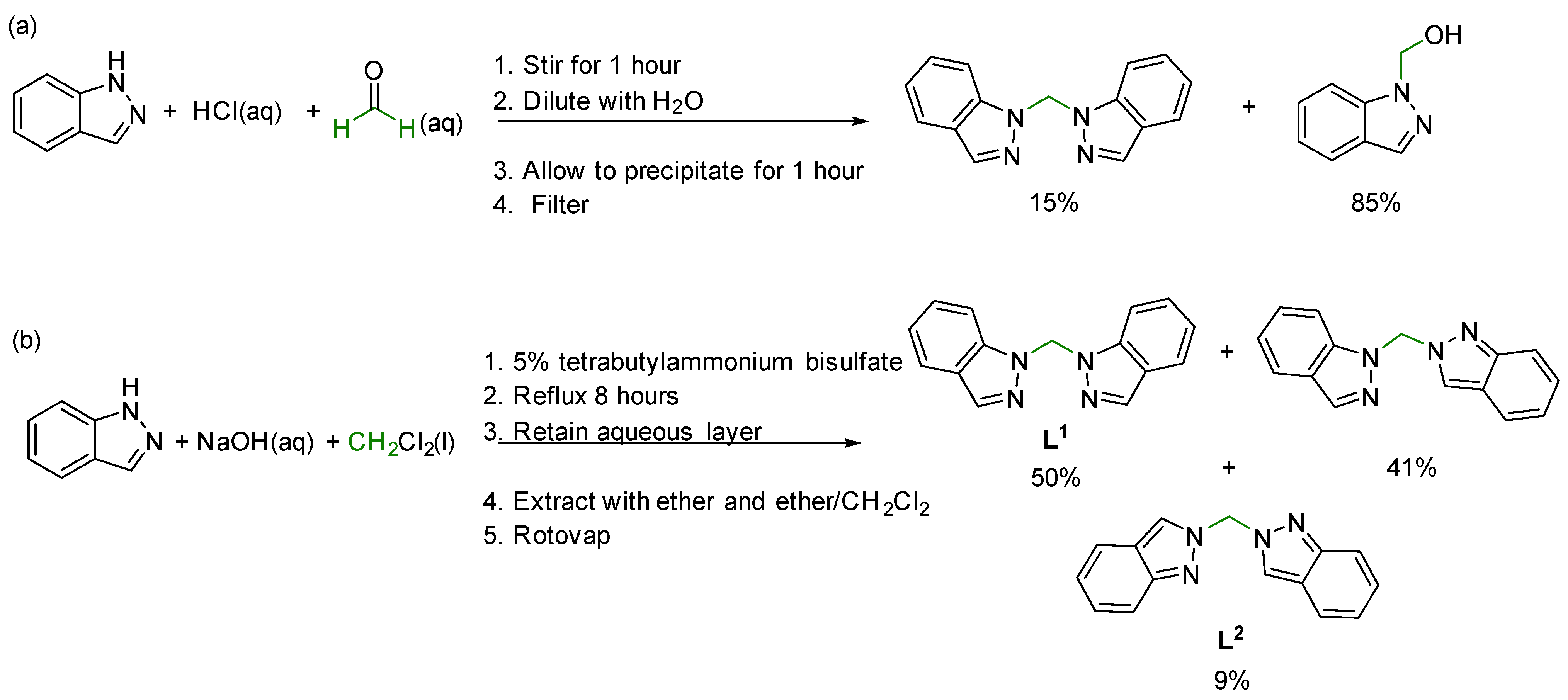
2.1. Synthesis and Characterization of Bis(1H-indazol-1-yl)methane, L1
2.1.1. General Procedure for the Formation of L1
2.1.2. Scale-Up Reaction Conditions
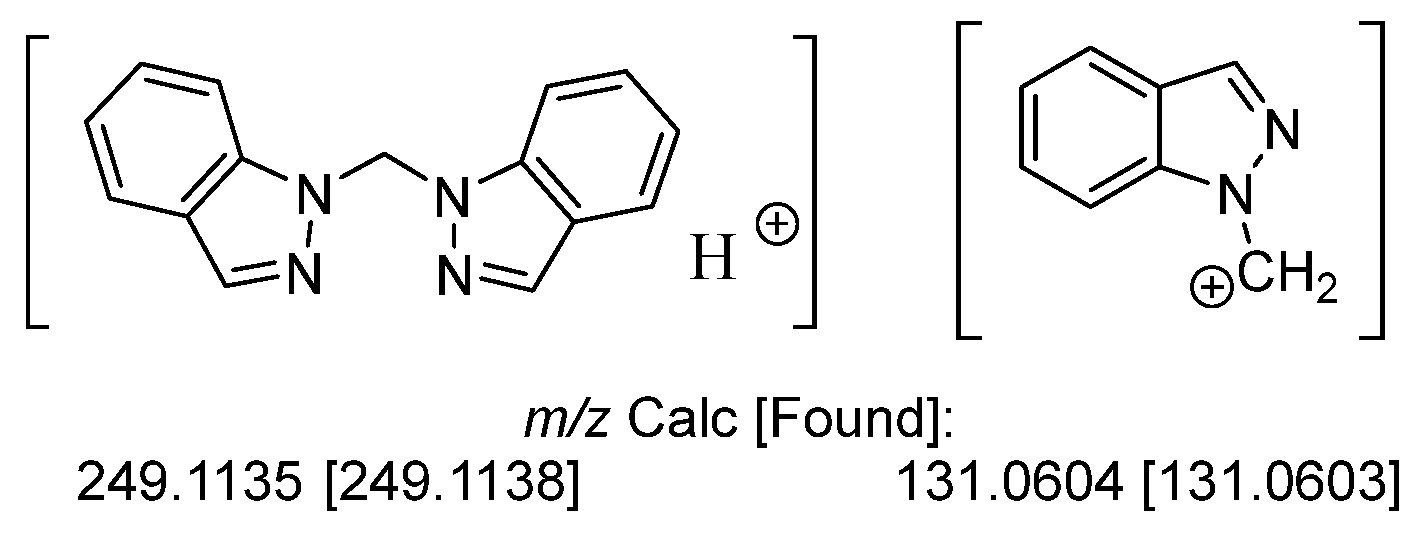
2.1.3. Characterization of L1
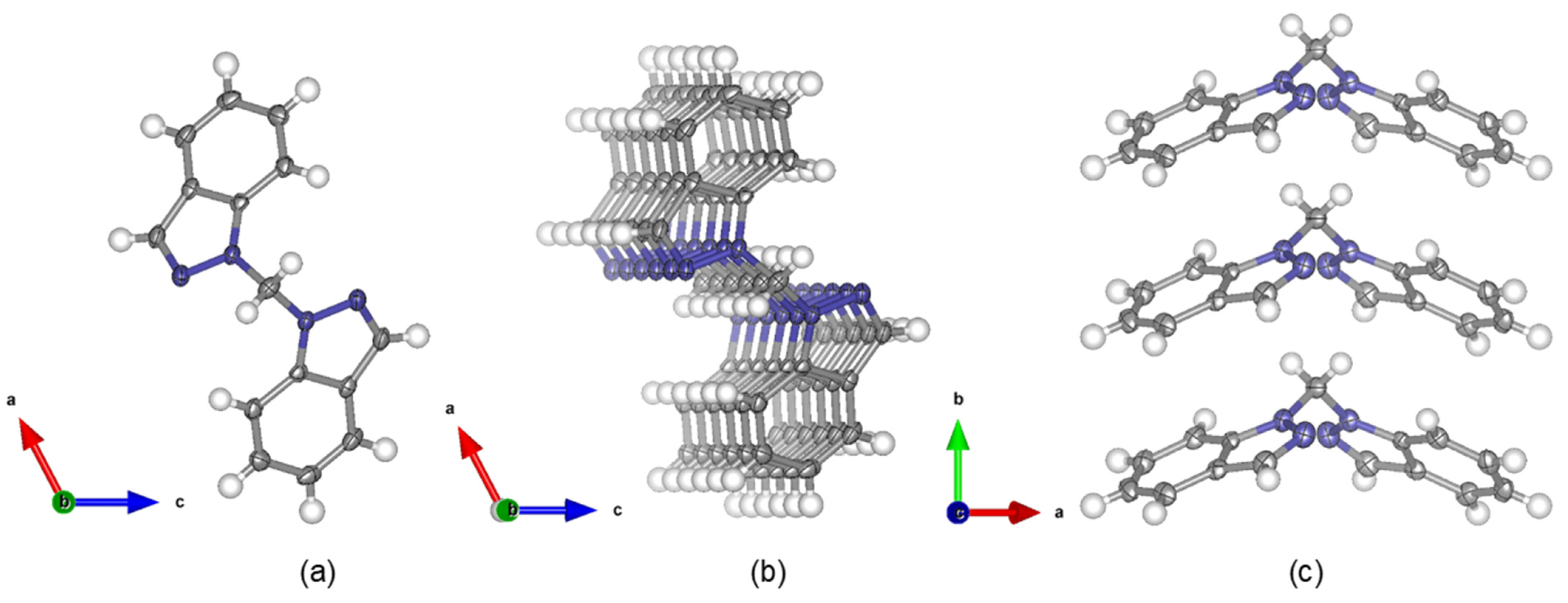
2.2. X-ray Crystallography
2.2.1. Experimental Conditions for Single Crystals Growth
2.2.2. X-ray Crystallography Data Collection
3. Results
3.1. One-Pot Synthesis of Bis(1H-indazol-1-yl)methane, L1
3.2. Characterization of Bis(1H-indazol-1-yl)methane, L1
4. Discussion
4.1. One-Pot Synthesis of Bis(1H-indazol-1-yl)methane, L1
4.2. Crystal Structure of Bis(1H-indazol-1-yl)methane, L1
4.3. Mechanistic Studies and Future Applications of L1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pettinari, C.; Pettinari, R. Metal derivatives of poly(pyrazolyl)alkanes☆II. Bis(pyrazolyl)alkanes and related systems. Coord. Chem. Rev. 2004, 249, 663–691. [Google Scholar] [CrossRef]
- Hückel, W.; Chneider, H.B. N-Tripyrazolyl-methan. Berichte Dtsch. Chem. Ges. A B Ser. 1937, 70, 2024–2026. [Google Scholar] [CrossRef]
- Alkorta, I.; Claramunt, R.M.; Díez-Barra, E.; Elguero, J.; de la Hoz, A.; López, C. The organic chemistry of poly(1H-pyrazol-1-yl)methanes. Coord. Chem. Rev. 2017, 339, 153–182. [Google Scholar] [CrossRef]
- Trofimenko, S. Geminal poly(1-pyrazolyl)alkanes and their coordination chemistry. J. Am. Chem. Soc. 1970, 92, 5118–5126. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Tejeda, J.; Sánchez-Barba, L.F. Recent Advances in the Design and Coordination Chemistry of Heteroscorpionate Ligands Bearing Stereogenic Centres. Eur. J. Inorg. Chem. 2008, 2008, 5309–5326. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Sánchez, A.L.; Sánchez-Barba, L.F. Metal complexes with heteroscorpionate ligands based on the bis(pyrazol-1-yl)methane moiety: Catalytic chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Pozharskii, F.T.; Kazanbieva, M.A.; Tertov, B.A. 1-Hydroxymethyl Derivates of Indazole. Zhurnal Obs. Khimii. 1964, 34, 3367–3370. [Google Scholar]
- Juliá, S.; Sala, P.; Del Mazo, J.; Sancho, M.; Ochoa, C.; Elguero, J.; Fayet, J.-P.; Vertut, M.-C. N-Polyazolylmethanes. 1. Synthesis and NMR Study of N,N′-Diazolylmethanes. J. Heterocycl. Chem. 1982, 19, 1141–1145. [Google Scholar] [CrossRef]
- Boryski, J. ChemInform Abstract: Acyclonucleosides of Indazole and Their Rearrangements. ChemInform 2010, 30, 1019–1027. [Google Scholar] [CrossRef]
- Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, W. Product Class 2: 1H- and 2H-Indazoles. In Science of Synthesis: Houben-Weyl Methods for Molecular Transformations; Neier, R., Ed.; Georg Thieme Verlag: New York, NY, USA, 2002; pp. 227–324. [Google Scholar]
- Pettinari, C.; Marinelli, A.; Marchetti, F.; Ngoune, J.; Galindo, A.; Álvarez, E.; Gómez, M. Synthesis and Coordination Chemistry of Two N2-Donor Chelating Di(indazolyl)methane Ligands: Structural Characterization and Comparison of Their Metal Chelation Aptitudes. Inorg. Chem. 2010, 49, 10543–10556. [Google Scholar] [CrossRef] [PubMed]
- Pettinari, C.; Marchetti, F.; Orbisaglia, S.; Pettinari, R.; Ngoune, J.; Gómez, M.; Santos, C.; Álvarez, E. Group 11 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane) ligands. CrystEngComm 2013, 15, 3892–3907. [Google Scholar] [CrossRef]
- Santos, C.; Gómez, M.; Álvarez, E.; Ngoune, J.; Marchetti, F.; Pettinari, R.; Pettinari, C. Group 9 and 10 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane ligands: Synthesis and structural characterization. New J. Chem. 2016, 40, 5695–5703. [Google Scholar] [CrossRef]
- Hart, K.F.; Joe, N.S.; Miller, R.M.; Nash, H.P.; Blake, D.J.; Morris, A.M. Synthesis and Characterization of trans-Dichlorotetrakis(imidazole)cobalt(III) Chloride: A New Cobalt(III) Coordination Complex with Potential Prodrug Properties. Bioinorg. Chem. Appl. 2018, 2018, 4560757. [Google Scholar] [CrossRef]
- Bruker. APEX4; Bruker AXS: Madison, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. SADABS—A Program for Area Detector Absorption Correction; University of Gottingen: Gottingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL, version 6.14; Bruker AXS: Madison, WI, USA, 1999. [Google Scholar]
- González, J.J.L.; Ureña, F.P.; Moreno, J.R.A.; Mata, I.; Molins, E.; Claramunt, R.M.; López, C.; Alkorta, I.; Elguero, J. The chiral structure of 1H-indazoles in the solid state: A crystallographic, vibrational circular dichroism and computational study. New J. Chem. 2012, 36, 749–758. [Google Scholar] [CrossRef]
- Mohammadi, B.; Khorrami, B.R. A simple and one-pot multi-component reaction to the synthesis of methylenebisamides. Mon. Für Chem.-Chem. Mon. 2017, 149, 1089–1092. [Google Scholar] [CrossRef]
- Patel, O.P.S.; Anand, D.; Maurya, R.K.; Yadav, P.P. H2O2/DMSO-Promoted Regioselective Synthesis of 3,3′-Bisimidazopyridinylmethanes via Intermolecular Oxidative C(sp2)–H Bond Activation of Imidazoheterocycles. J. Org. Chem. 2016, 81, 7626–7634. [Google Scholar] [CrossRef]
- Ai, H.-J.; Qi, X.; Peng, J.-B.; Ying, J.; Wu, X.-F. Palladium-Catalyzed Cross-Coupling of Arylboronic Acid and Benzonitriles Using DMSO as the Methylene Source. Asian J. Org. Chem. 2018, 7, 2045–2048. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Tavakoli, G.; Saeednia, B. Ni-Catalyzed Synthesis of Methylenebisamides: Dual Role of DMSO Both as Methylene Source and Oxidant. ChemistrySelect 2017, 2, 1316–1322. [Google Scholar] [CrossRef]
- Zheng, K.; Zhuang, S.; Shu, W.; Wu, Y.; Yang, C.; Wu, A. Molecular iodine-mediated formal [2+1+1+1] cycloaddition access to pyrrolo[2,1-a]isoquinolines with DMSO as the methylene source. Chem. Commun. 2018, 54, 11897–11900. [Google Scholar] [CrossRef]
- Tashrifi, Z.; Khanaposhtani, M.M.; Larijani, B.; Mahdavi, M. Dimethyl Sulfoxide: Yesterday’s Solvent, Today’s Reagent. Adv. Synth. Catal. 2020, 362, 65–86. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Shen, Z.; Yuan, Y.; Sun, P. Synthesis of symmetrical methylene-bridged imidazoheterocycles using DMSO as methylene source under metal-free conditions. Org. Biomol. Chem. 2016, 14, 6523–6530. [Google Scholar] [CrossRef]
- Catalan, J.; de Paz, J.; Elguero, L.G. Importance of aromaticity on the relative stabilities of indazole annular tautomers: An ab initio study. J. Chem. Soc. Perkin Trans. 1996, 2, 57–60. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Theoretical estimation of the annular tautomerism of indazoles. J. Phys. Org. Chem. 2005, 18, 719–724. [Google Scholar] [CrossRef]
- Churchill, M.R.; Churchill, D.G.; Huynh, M.H.V.; Takeuchi, K.J.; Castellano, R.K.; Jameson, D.L. Crystal and molecular structure of di(1-pyrazolyl)methane, CH2(C3N2H3)2. J. Chem. Crystallogr. 1996, 26, 93–97. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Larsen, F.K.; Lehmann, M.S.; Søtofte, I.; Rasmussen, S.E.; Shimizu, A. A Neutron Diffraction Study of the Crystal and Molecular Structure of Pyrazole, C3H4N2. Acta Chem. Scand. 1970, 24, 3248–3258. [Google Scholar] [CrossRef] [Green Version]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | L1 |
|---|---|
| Formula | C15H12N4 |
| Formula Weight (g/mol) | 248.29 |
| Temperature | 100(2) K |
| Crystal system | Monoclinic |
| space group, Z | C2, 4 |
| a (Å) | 23.6129(19) |
| b (Å) | 4.0856(4) |
| c (Å) | 13.9334(12) |
| α (°) | 90 |
| β (°) | 117.743(5) |
| γ (°) | 90 |
| Volume (Å3) | 1189.67(19) |
| (g/cm3) | 1.3806 |
| R1a | 0.0863 |
| wR2 a | 0.0909 |
| GOF | 1.037 |
| Entry | Time (h) | Metal Catalyst | Additive | Dimer Formation? | Isolated Yield a |
|---|---|---|---|---|---|
| 1 | 24 | - | - | Partial | - |
| 2 | 72 | - | - | Yes b | - b |
| 3 | 24 | - | H2O2 | No | - |
| 4 | 24 | 10% CoCl2·6H2O | - | Yes | 94% |
| 5 | 24 | 1% CoCl2·6H2O | - | Yes | 96% |
| 6 | 24 | 1% CoCl2·6H2O | H2O2 | No | - |
| 7 | 24 | 1% CoCl2 | - | Yes | 92% |
| 8 | 24 | 1% [Co(pyridine)4Cl2]Cl | - | Yes | 96% |
| 9 | 24 | 1% FeCl2·4H2O | - | Yes | 98% |
| 10 | 24 | 1% FeCl2 | - | Yes | 96% |
| 11 | 24 | 1% NiCl2·6H2O | - | Yes | 96% |
| 12 | 24 | 1% ZnCl2 | - | Yes | 94% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lind, N.M.; Joe, N.S.; Newell, B.S.; Morris, A.M. High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts. Reactions 2022, 3, 59-69. https://doi.org/10.3390/reactions3010005
Lind NM, Joe NS, Newell BS, Morris AM. High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts. Reactions. 2022; 3(1):59-69. https://doi.org/10.3390/reactions3010005
Chicago/Turabian StyleLind, Natalie M., Natalie S. Joe, Brian S. Newell, and Aimee M. Morris. 2022. "High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts" Reactions 3, no. 1: 59-69. https://doi.org/10.3390/reactions3010005
APA StyleLind, N. M., Joe, N. S., Newell, B. S., & Morris, A. M. (2022). High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts. Reactions, 3(1), 59-69. https://doi.org/10.3390/reactions3010005