Understanding Conformational Polymorphism in Ganciclovir: A Holistic Approach




Abstract
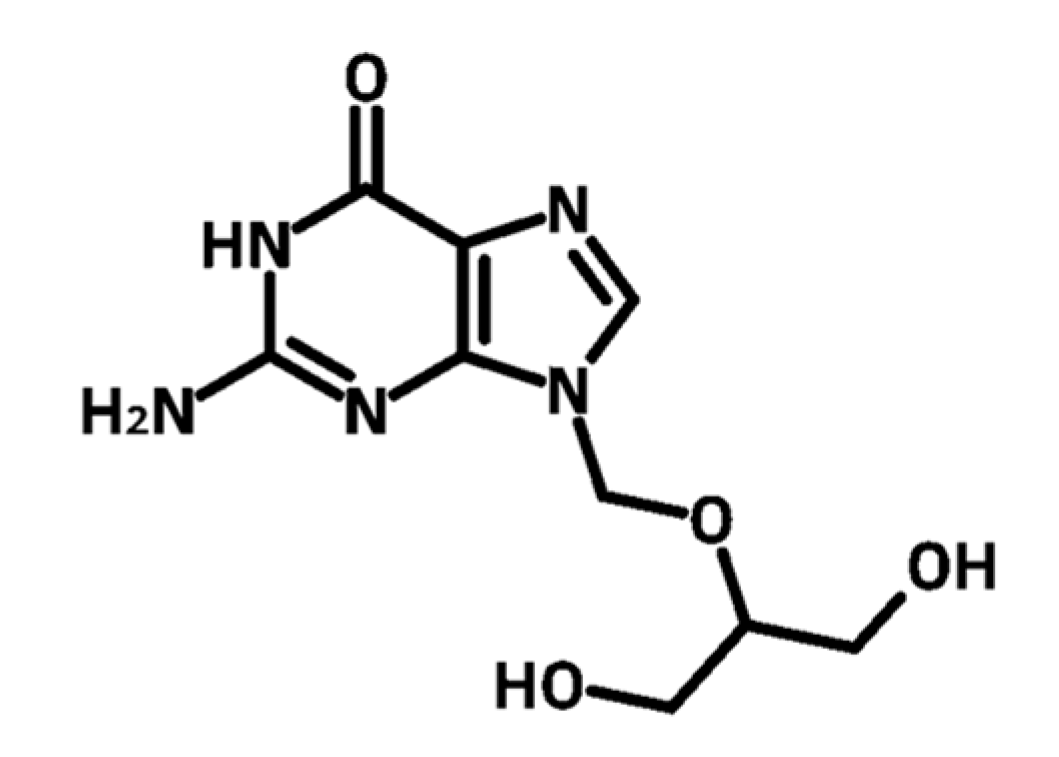
:1. Introduction
2. Computational Methods
2.1. Non-Bonding Interactions Analysis
2.2. Energy Framework Analysis
2.3. Polymorph Assessment through Hydrogen Bond Propensity Models
3. Results and Discussion
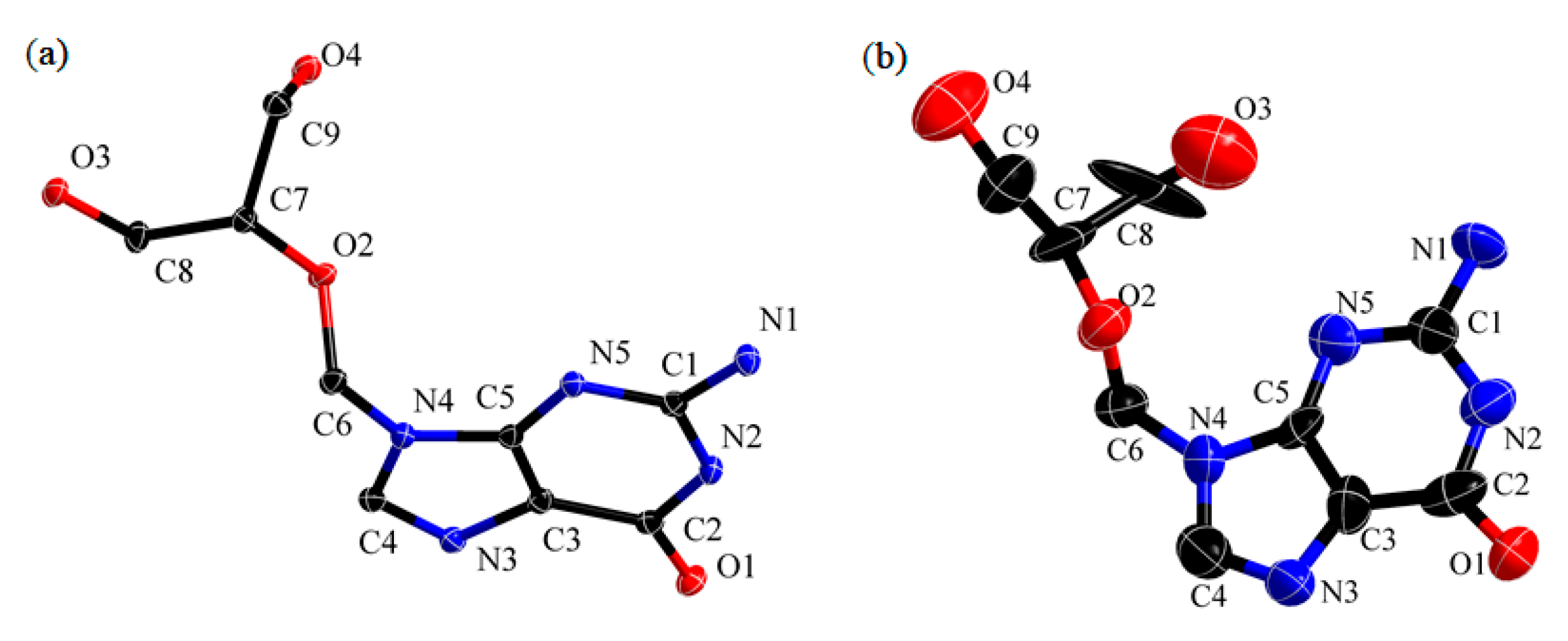
3.1. Intramolecular Level
3.2. Intermolecular Level
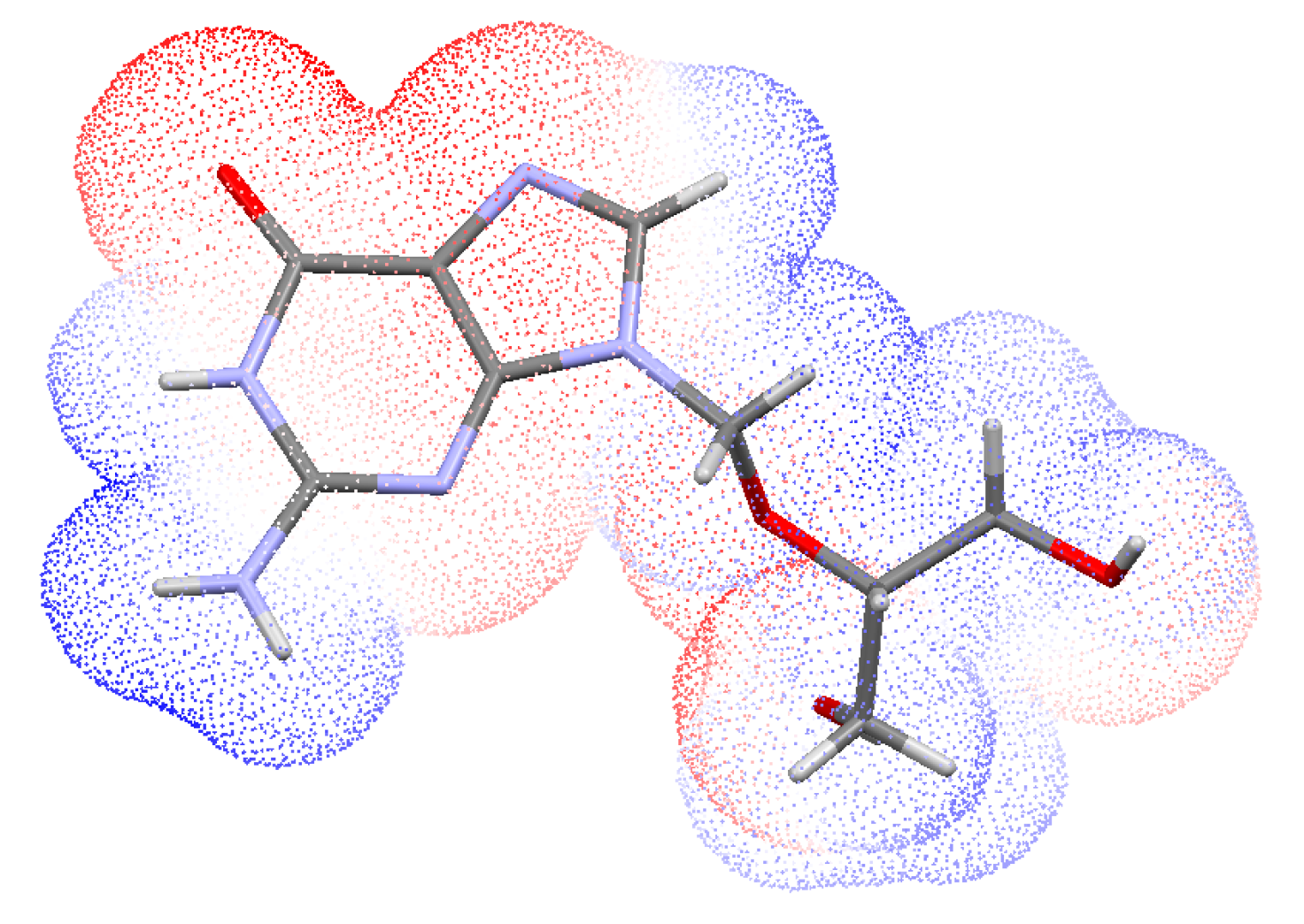
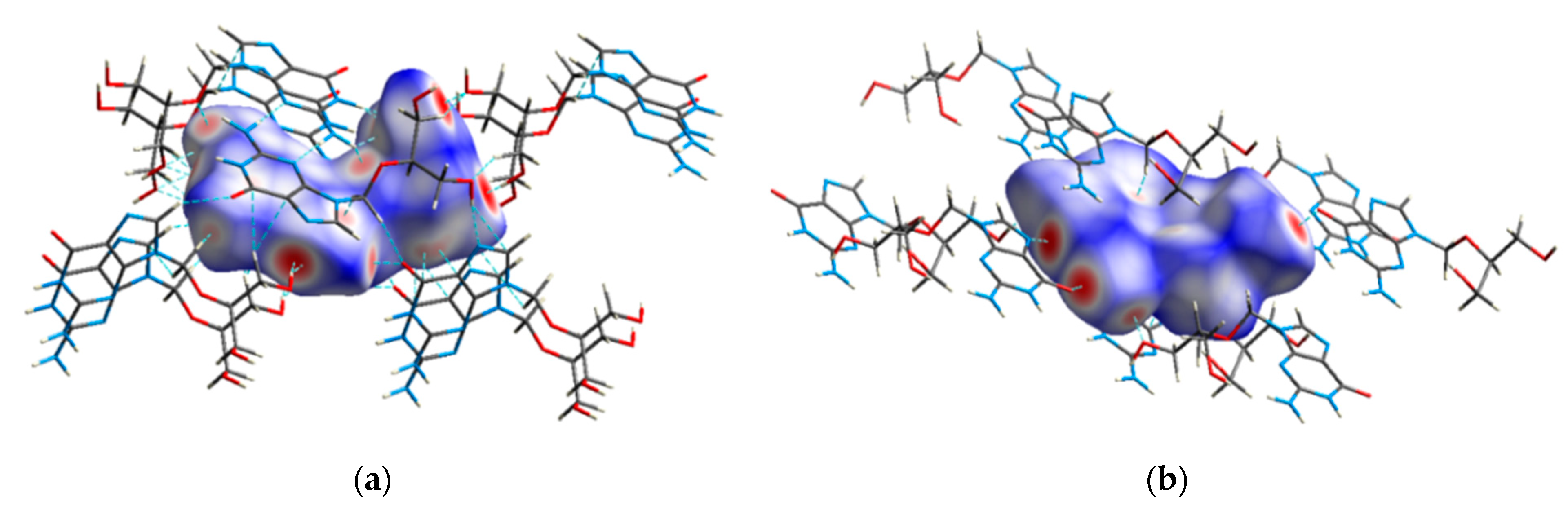
3.3. Hirshfeld Surface Analysis
3.4. Supramolecular Analysis
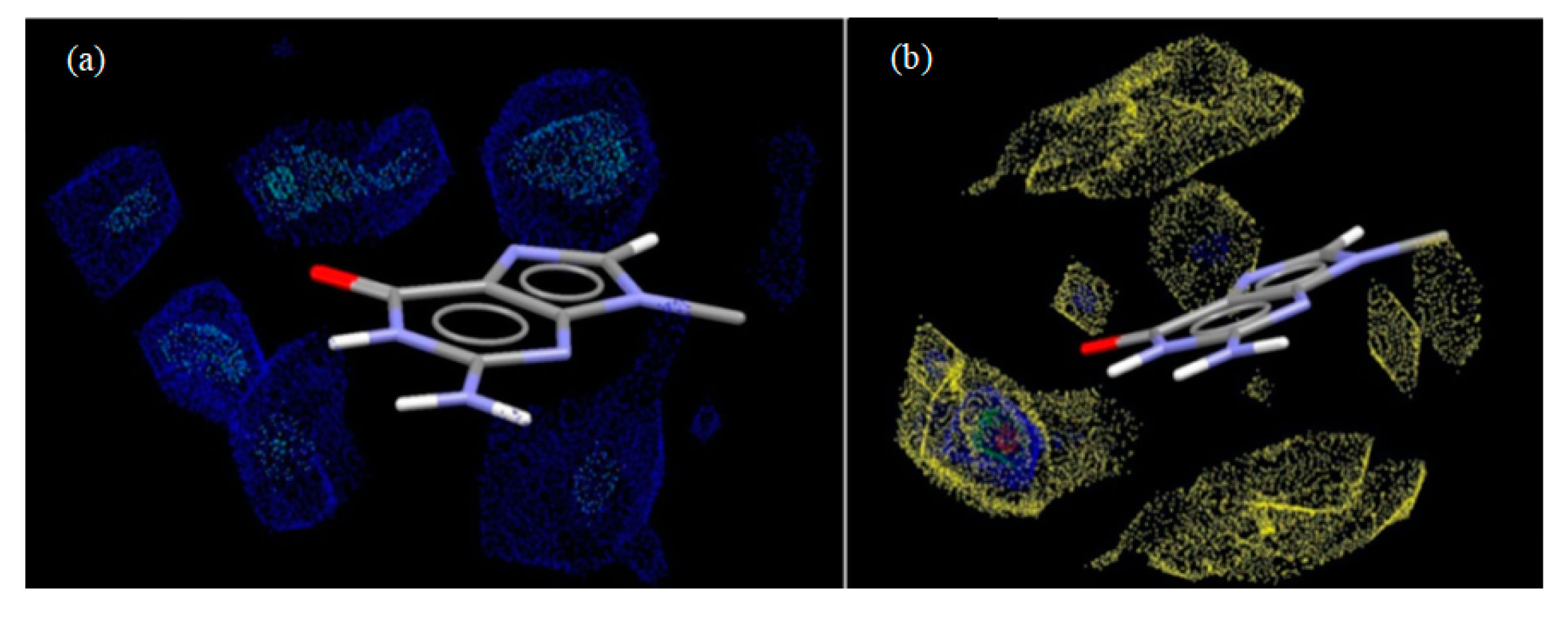
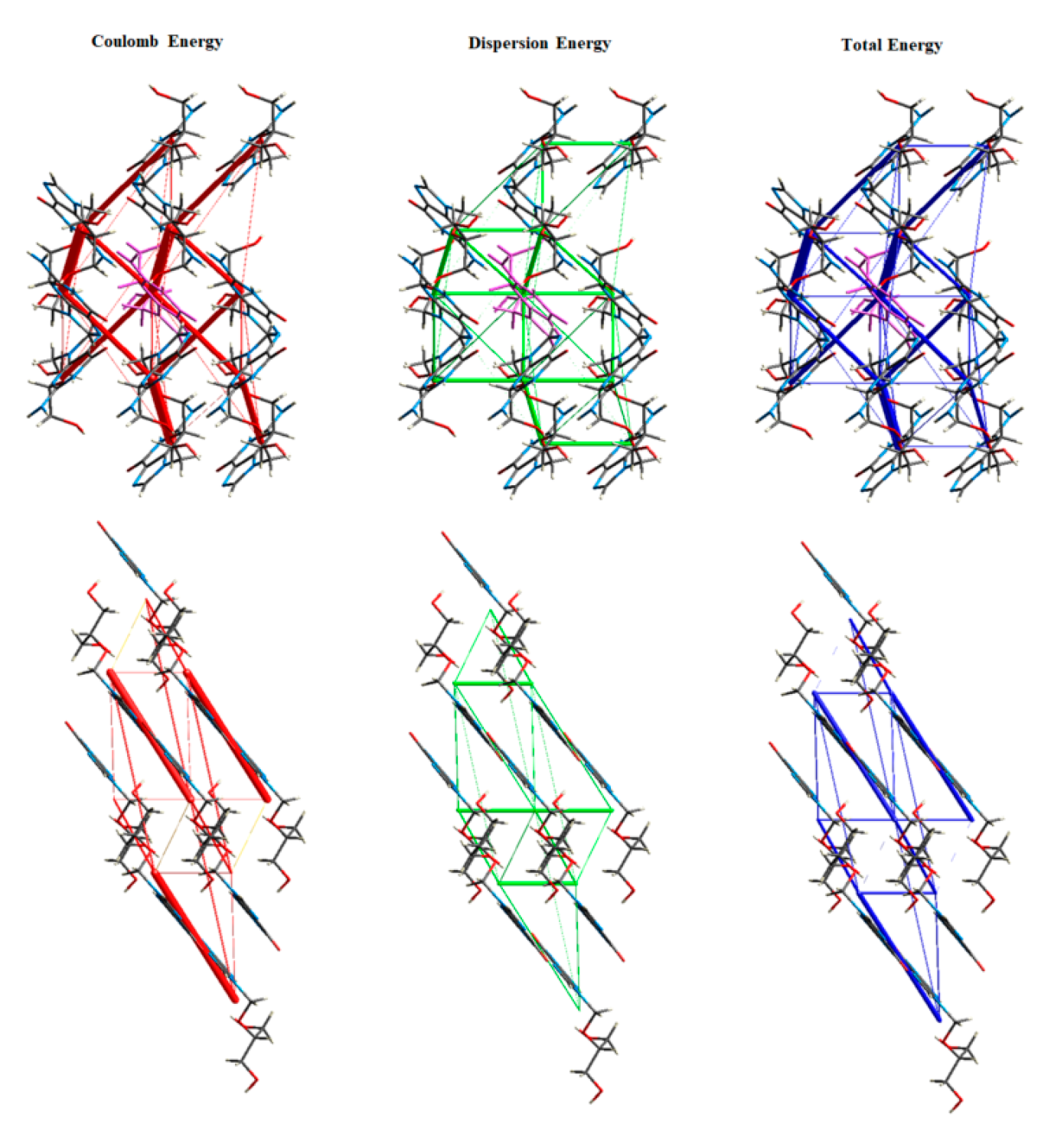
3.5. Energy Frameworks
3.6. Polymorph Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salazar, D.E.; Gormley, G. Modern Drug Discovery and Development. In Clinical and Translational Science, 2nd ed.; Robertson, D., Williams, G.H., Eds.; Academic Press: Nashville, TN, USA, 2017; pp. 719–743. [Google Scholar]
- Koenig, S.G.; Leahy, D.K.; Wells, A.S. Evaluating the Impact of a Decade of Funding from the Green Chemistry Institute Pharmaceutical Roundtable. Org. Process Res. Dev. 2018, 22, 1344–1359. [Google Scholar] [CrossRef]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nature Rev. Drug Discov. 2004, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. Computed Crystal Energy Landscapes for Understanding and Predicting Organic Crystal Structures and Polymorphism. Acc. Chem. Res. 2009, 42, 117–126. [Google Scholar] [CrossRef]
- Abramov, Y.A. Current Computational Approaches to Support Pharmaceutical Solid Form Selection. Org. Process Res. Dev. 2013, 17, 472–485. [Google Scholar] [CrossRef]
- Sandhu, B.; McLean, A.; Sinha, A.S.; Desper, J.; Sarjeant, A.A.; Vyas, S.; Reutzel-Edens, S.M.; Aakeröy, C.B. Evaluating Competing Intermolecular Interactions through Molecular Electrostatic Potentials and Hydrogen-Bond Propensities. Cryst. Growth Des. 2018, 18, 466–478. [Google Scholar] [CrossRef]
- Spiteri, L.; Baisch, U.; Vella-Zarb, L. Correlations and statistical analysis of solvent molecule hydrogen bonding—A case study of dimethyl sulfoxide (DMSO). CrystEngComm 2018, 20, 1291–1303. [Google Scholar] [CrossRef]
- Thomas, S.P.; Spackman, M. The Polymorphs of ROY: A Computational Study of Lattice Energies and Conformational Energy Differences*. Aust. J. Chem. 2018, 71. [Google Scholar] [CrossRef] [Green Version]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Verheyden, J.P.; Martin, J.C. 9-(1,3-dihydroxy-2-propoxymethyl)-guanine as an Antiviral. U.S. Patent No. 4355032, 19 October 1982. [Google Scholar]
- Mallipeddi, S.M.; Sterling, J.E.; Koneru, P.B. Ganciclovir Compositions and Related Methods. U.S. Patent No. 9486530, 8 November 2016. [Google Scholar]
- Parr, A.; Hidalgo, I.J.; Bode, C.; Brown, W.; Yazdanian, M.; Gonzalez, M.A.; Sagawa, K.; Miller, K.; Jiang, W.; Stippler, E.S. The Effect of Excipients on the Permeability of BCS Class III Compounds and Implications for Biowaivers. Pharm. Res. 2016, 33, 167–176. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Drug Information 2017. WHO Drug Inf. 2017, 31, 385–492. Available online: https://apps.who.int/iris/handle/10665/330952 (accessed on 26 January 2021).
- Sarbajna, R.; Anil, P.; Sivalakshmi Devi, A.; Suryanarayana, M.V.; Sethi, M.; Dutta, D. Studies on Crystal Modifications of Ganciclovir; Taylor & Francis: London, UK, 2011; Volume 537, pp. 141–154. [Google Scholar]
- Kawamura, T.; Hirayama, N. Crystal Structure of Ganciclovir. X-Ray Struct. Anal. Online 2009, 25, 51–52. [Google Scholar] [CrossRef] [Green Version]
- Roque-Flores, R.L.; Guzei, I.A.; Matos, J.d.R.; Yu, L. Polymorphs of the antiviral drug ganciclovir. Acta Crystallogr. Sect. C 2017, 73, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Roque-Flores, R.L.; Matos, J.d.R. Simultaneous measurements of X-ray diffraction–differential scanning calorimetry. J. Therm. Anal. Calorim. 2019, 137, 1347–1358. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Diamond—Crystal and Molecular Structure Visualisation, 4.6.4; Crystal Impact: Bonn, Germany, 2020. [Google Scholar]
- Galek, P.T.A.; Pidcock, E.; Wood, P.A.; Bruno, I.J.; Groom, C.R. One in half a million: A solid form informatics study of a pharmaceutical crystal structure. CrystEngComm 2012, 14, 2391–2403. [Google Scholar] [CrossRef]
- Lewars, E.G. Semiempirical calculations. In Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Hingham, MA, USA, 2016. [Google Scholar]
- Rocha Gerd, B.; Freire Ricardo, O.; Simas Alfredo, M.; Stewart James, J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Bruno, I.; Cole, J.; Lommerse, J.; Rowland, R.; Taylor, R.; Verdonk, M. IsoStar: A library of information about nonbonded interactions. J. Comput. Aided Mol. Design 1997, 11, 525–537. [Google Scholar] [CrossRef]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. CrystEngComm 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Bruno Ian, J.; Cole Jason, C.; Edgington Paul, R.; Kessler, M.; Macrae Clare, F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B 2008, 58, 389–397. [Google Scholar] [CrossRef]
- Wood, P.A.; Allen, F.H.; Pidcock, E. Hydrogen-bond directionality at the donor H atom—Analysis of interaction energies and database statistics. CrystEngComm 2009, 11, 1563–1571. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Cole, J.; Korb, O.; McCabe, P. Knowledge-Based Libraries for Predicting the Geometric Preferences of Druglike Molecules. J. Chem. Inf. Modeling 2014, 54, 2500–2514. [Google Scholar] [CrossRef]
- Fernandes, J.A.; Galli, S.; Palmisano, G.; Volante, P.; Mendes, R.F.; Paz, F.A.A.; Masciocchi, N. Reviewing the Manifold Aspects of Ganciclovir Crystal Forms. Cryst. Growth Design 2016, 16, 4108–4118. [Google Scholar] [CrossRef]
- Scheffer, J.; Wong, Y.F.; Patil, A.O.; Curtin, D.Y.; Paul, I.C. CPMAS (cross-polarization magic angle spinning) carbon-13 NMR spectra of quinones, hydroquinones, and their complexes. Use of CMR to follow a reaction in the solid state. J. Am. Chem. Soc. 1985, 107, 4898–4904. [Google Scholar] [CrossRef]
- Steiner, T. C–H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent inter-action (NCI) plots and the calculation of inter-action energies in the analysis of mol-ecular packing. Acta Cryst. E Cryst. Commun. 2019, 75, 308–318. [Google Scholar] [CrossRef] [Green Version]
- Vella-Zarb, L.; Dinnebier, R.E.; Baisch, U. The Devil is in the Detail: A Rare H-Bonding Motif in New Forms of Docetaxel. Cryst. Growth Design 2013, 13, 4402–4410. [Google Scholar] [CrossRef]
- Matta, C.F.; Hernández-Trujillo, J.; Tang, T.-H.; Bader, R.F.W. Hydrogen–Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals. Chem. A Eur. J. 2003, 9, 1940–1951. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Fabbiani, F.P.A.; Spackman, M.A. Comparison of Polymorphic Molecular Crystal Structures through Hirshfeld Surface Analysis. Cryst. Growth Design 2007, 7, 755–769. [Google Scholar] [CrossRef]
- Carugo, O.; Blatova, O.A.; Medrish, E.O.; Blatov, V.A.; Proserpio, D.M. Packing topology in crystals of proteins and small molecules: A comparison. Sci. Rep. 2017, 7, 13209. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.-F.; Wang, J.-Y.; Pei, J. Control of π–π Stacking via Crystal Engineering in Organic Conjugated Small Molecule Crystals. Cryst. Growth Design 2018, 18, 7–15. [Google Scholar] [CrossRef]
- Yu, L. Inferring thermodynamic stability relationship of polymorphs from melting data. J. Pharm. Sci. 1995, 84, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Le, C.T. Applied Categorical Data Analysis and Translational Research; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Forte, R.M. Mastering Predictive Analytics with R; Packt Publishing: Birmingham, UK, 2015. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form I | Form II | |
|---|---|---|
| Volume (Å3) | 250.18 | 268.07 |
| Packing coefficient | 0.770978 | 0.725452 |
| Density (g/cm3) | 1.653 | 1.549 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiteri, L.; Baisch, U.; Vella-Zarb, L. Understanding Conformational Polymorphism in Ganciclovir: A Holistic Approach. Chemistry 2021, 3, 126-137. https://doi.org/10.3390/chemistry3010010
Spiteri L, Baisch U, Vella-Zarb L. Understanding Conformational Polymorphism in Ganciclovir: A Holistic Approach. Chemistry. 2021; 3(1):126-137. https://doi.org/10.3390/chemistry3010010
Chicago/Turabian StyleSpiteri, Lorella, Ulrich Baisch, and Liana Vella-Zarb. 2021. "Understanding Conformational Polymorphism in Ganciclovir: A Holistic Approach" Chemistry 3, no. 1: 126-137. https://doi.org/10.3390/chemistry3010010
APA StyleSpiteri, L., Baisch, U., & Vella-Zarb, L. (2021). Understanding Conformational Polymorphism in Ganciclovir: A Holistic Approach. Chemistry, 3(1), 126-137. https://doi.org/10.3390/chemistry3010010