
Structural Diversity of Lithium Oligo-α-Pyridylamides



Abstract
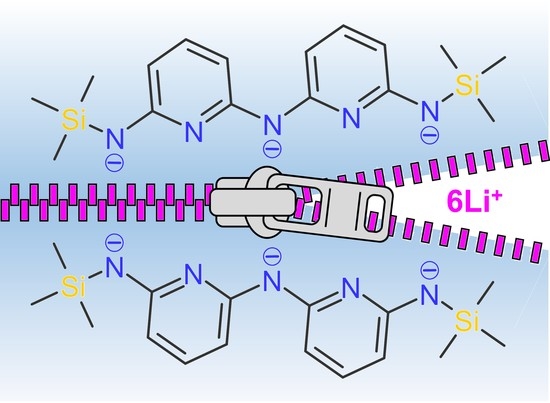
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. H5A and H3L
3.2. [Li6L2(thf)6]·C6H14 (1·C6H14)
3.3. [Li6L2(thf)4] (2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chlupatý, T.; Růžička, A. Hybrid amidinates and guanidinates of main group metals. Coord. Chem. Rev. 2016, 314, 103–113. [Google Scholar] [CrossRef]
- Zechovský, J.; Mrózek, O.; Samsonov, M.; Jambor, R.; Růžička, A.; Dostál, L. Coordination capabilities of bis-(2-pyridyl)amides in the field of divalent germanium, tin and lead compounds. Dalton Trans. 2021, 50, 6321–6332. [Google Scholar] [CrossRef]
- Kempe, R. The Strained η2-NAmido−NPyridine Coordination of Aminopyridinato Ligands. Eur. J. Inorg. Chem. 2003, 2003, 791–803. [Google Scholar] [CrossRef]
- Berry, J.F.; Cotton, F.A.; Daniels, L.M.; Murillo, C.A.; Wang, X. Oxidation of Ni3(dpa)4Cl2 and Cu3(dpa)4Cl2: Nickel−Nickel Bonding Interaction, but No Copper−Copper Bonds. Inorg. Chem. 2003, 42, 2418–2427. [Google Scholar] [CrossRef]
- Chen, P.-J.; Sigrist, M.; Horng, E.-C.; Lin, G.-M.; Lee, G.-H.; Chen, C.-h.; Peng, S.-M. A ligand design with a modified naphthyridylamide for achieving the longest EMACs: The 1st single-molecule conductance of an undeca-nickel metal string. Chem. Commun. 2017, 53, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Otsubo, K.; Yoshida, Y.; Kimura, Y.; Sugimoto, K.; Kitagawa, H. Synthesis and Magnetic Properties of a Dimerized Trinuclear Ni String Complex, [Ni6Cl2(dpa)8](I5)2·0.25I2 (dpa– = 2,2′-Dipyridylamide Anion). Inorg. Chem. 2021, 60, 16029–16034. [Google Scholar] [CrossRef]
- Guillet, G.L.; Arpin, K.Y.; Boltin, A.M.; Gordon, J.B.; Rave, J.A.; Hillesheim, P.C. Synthesis and Characterization of a Linear Triiron(II) Extended Metal Atom Chain Complex with Fe–Fe Bonds. Inorg. Chem. 2020, 59, 11238–11243. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Wang, X. Trinuclear complexes of copper, cobalt and iron with N,N′-di(2-pyridyl) formamidinate ligands, [M3(DPyF)4][PF6]2. Inorg. Chem. Commun. 1998, 1, 281–283. [Google Scholar] [CrossRef]
- Srinivasan, A.; Musgrave, R.A.; Rouzières, M.; Clérac, R.; McGrady, J.E.; Hillard, E.A. A linear metal–metal bonded tri-iron single-molecule magnet. Chem. Commun. 2021, 57, 13357–13360. [Google Scholar] [CrossRef]
- Brogden, D.W.; Berry, J.F. Coordination Chemistry of 2,2’-Dipyridylamine: The Gift That Keeps on Giving. Comments Inorg. Chem. 2016, 36, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.-A.; Cheng, M.-C.; Chen, C.-h.; Peng, S.-M. From Homonuclear Metal String Complexes to Heteronuclear Metal String Complexes. Eur. J. Inorg. Chem. 2015, 2015, 2510–2523. [Google Scholar] [CrossRef]
- Berry, J.F.; Cotton, F.A.; Murillo, C.A.; Chan, Z.-K.; Yeh, C.-W.; Chen, J.-D. Linear Trichromium, Tricobalt, Trinickel, and Tricopper Complexes of 2,2′-Dipyridylamide. Inorg. Synth. 2014, 36, 103–110. [Google Scholar] [CrossRef]
- Hurley, T.J.; Robinson, M.A. Nickel(II)-2,2’-dipyridylamine system. I. Synthesis and stereochemistry of the complexes. Inorg. Chem. 1968, 7, 33–38. [Google Scholar] [CrossRef]
- Yang, E.-C.; Cheng, M.-C.; Tsai, M.-S.; Peng, S.-M. Structure of a linear unsymmetrical trinuclear cobalt(II) complex with a localized CoII–CoII bond: Dichlorotetrakis[µ3-bis(2-pyridyl)amido]tricobalt(II). J. Chem. Soc. Chem. Commun. 1994, 2377–2378. [Google Scholar] [CrossRef]
- Cotton, F.A.; Daniels, L.M.; Murillo, C.A.; Pascual, I. Compounds with linear, bonded trichromium chains. J. Am. Chem. Soc. 1997, 119, 10223–10224. [Google Scholar] [CrossRef]
- Nicolini, A.; Galavotti, R.; Barra, A.-L.; Borsari, M.; Caleffi, M.; Luo, G.; Novitchi, G.; Park, K.; Ranieri, A.; Rigamonti, L.; et al. Filling the Gap in Extended Metal Atom Chains: Ferromagnetic Interactions in a Tetrairon(II) String Supported by Oligo-α-pyridylamido Ligands. Inorg. Chem. 2018, 57, 5438–5448. [Google Scholar] [CrossRef]
- Nicolini, A.; Affronte, M.; SantaLucia, D.J.; Borsari, M.; Cahier, B.; Caleffi, M.; Ranieri, A.; Berry, J.F.; Cornia, A. Tetrairon(II) extended metal atom chains as single-molecule magnets. Dalton Trans. 2021, 50, 7571–7589. [Google Scholar] [CrossRef]
- Chipman, J.A.; Berry, J.F. Paramagnetic Metal–Metal Bonded Heterometallic Complexes. Chem. Rev. 2020, 120, 2409–2447. [Google Scholar] [CrossRef]
- Glatz, G.; Kempe, R. Crystal structure of bis-μ3(N,N’-bis-(trimethylsilyl)-pyridine-2-amine-6-amido)-μ4(N,N’-bis-(trimethylsilyl)-pyridine-2,6-diamido)-di-tetrahydrofuran-tetralithium, [(C11H21N3Si2)(C11H22N3Si2)2(C4H8O)2Li4]. Z. Kristallogr. New Cryst. Struct. 2008, 223, 307–308. [Google Scholar] [CrossRef]
- Skvortsov, G.G.; Fukin, G.K.; Ketkov, S.Y.; Cherkasov, A.V.; Lyssenko, K.A.; Trifonov, A.A. Benzonitrile Insertion into Silylarylamides—ansa-Bis(benzamidinate) Ligand Systems with Rigid o- and m-Phenylene Linkers in the Synthesis of Lithium and Rare Earth Complexes. Eur. J. Inorg. Chem. 2013, 2013, 4173–4183. [Google Scholar] [CrossRef]
- Fairley, M.; Bole, L.J.; Mulks, F.F.; Main, L.; Kennedy, A.R.; O’Hara, C.T.; García-Alvarez, J.; Hevia, E. Ultrafast amidation of esters using lithium amides under aerobic ambient temperature conditions in sustainable solvents. Chem. Sci. 2020, 11, 6500–6509. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Elsevier, Ed.; Butterworth-Heinemann: Burlington, VT, USA, 2003; ISBN 0-7506-7571-3. [Google Scholar]
- den Hertog, H.J.; Wibaut, J.P. On the reactivity of bromine atoms in brominated pyridines. Preparation of some 2:6-disubstitution products of pyridine. Recl. Trav. Chim. Pays-Bas 1936, 55, 122–130. [Google Scholar] [CrossRef]
- Love, B.E.; Jones, E.G. The Use of Salicylaldehyde Phenylhydrazone as an Indicator for the Titration of Organometallic Reagents. J. Org. Chem. 1999, 64, 3755–3756. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, C.A.; Matuszak, A.J. Readily available anhydrous ether solutions of hydrogen chloride. J. Chem. Educ. 1967, 44, 108. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.H.; Chen, A.D.; Johnson, C.S. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Bachmann, S.; Neufeld, R.; Dzemski, M.; Stalke, D. New External Calibration Curves (ECCs) for the Estimation of Molecular Weights in Various Common NMR Solvents. Chem. Eur. J. 2016, 22, 8462–8465. [Google Scholar] [CrossRef]
- Bachmann, S.; Gernert, B.; Stalke, D. Solution structures of alkali metal cyclopentadienides in THF estimated by ECC-DOSY NMR-spectroscopy (incl. software). Chem. Commun. 2016, 52, 12861–12864. [Google Scholar] [CrossRef] [Green Version]
- APEX2, SADABS, SAINT; Bruker-AXS, Inc.: Madison, WI, USA, 2012.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR 92—A program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced rigid-bond restraints. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef] [Green Version]
- Persistence of Vision Raytracer (Version 3.7) [Computer software]; Persistence of Vision Pty. Ltd.: Williamstown, Australia, 2013; Available online: http://www.povray.org/download/ (accessed on 24 May 2022).
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, J.; Stearns, B.; Dexter, M.; Lott, W.A.I. Derivatives of Aminopyridines. J. Am. Chem. Soc. 1947, 69, 1147–1150. [Google Scholar] [CrossRef]
- Bernstein, J.; Stearns, B.; Shaw, E.; Lott, W.A., II. Derivatives of 2,6-Diaminopyridine. J. Am. Chem. Soc. 1947, 69, 1151–1158. [Google Scholar] [CrossRef]
- Mibu, N.; Yokomizo, K.; Miyata, T.; Sumoto, K. N-Monocarbamoyl Derivatives of Symmetrical Diamines with Antiviral Activity. Chem. Pharm. Bull. 2007, 55, 1406–1411. [Google Scholar] [CrossRef] [Green Version]
- Schrage, B.R.; Chanawanno, K.; Crandall, L.A.; Ziegler, C.J. The synthesis of a hexameric expanded hemiporphyrazine. J. Porphyr. Phthalocyanines 2020, 24, 129–134. [Google Scholar] [CrossRef]
- Lemport, P.S.; Ostapchuk, P.N.; Bobrikova, A.A.; Petrovskii, P.V.; Kagramanov, N.D.; Bodrin, G.V.; Nifant’ev, E.E. Direct phosphorylation in the synthesis of new bis(phosphorylamino)pyridine ligands. Mendeleev Commun. 2010, 20, 223–225. [Google Scholar] [CrossRef]
- Wang, W.-Z.; Ismayilov, R.H.; Lee, G.-H.; Liu, I.P.-C.; Yeh, C.-Y.; Peng, S.-M. The nano-scale molecule with the longest delocalized metal–metal bonds: Linear heptacobalt(II) metal string complexes [Co7(µ7-L)4X2]. Dalton Trans. 2007, 830–839. [Google Scholar] [CrossRef]
- Beer, L.; Britten, J.F.; Brusso, J.L.; Cordes, A.W.; Haddon, R.C.; Itkis, M.E.; MacGregor, D.S.; Oakley, R.T.; Reed, R.W.; Robertson, C.M. Prototypal Dithiazolodithiazolyl Radicals: Synthesis, Structures, and Transport Properties. J. Am. Chem. Soc. 2003, 125, 14394–14403. [Google Scholar] [CrossRef]
- Deacon, G.B.; Forsyth, C.M.; Scott, N.M. Structurally diverse organoamides and organoamido-, organometallic-lithium aggregates from reactions of N-(2-phenoxyphenyl)-N-(trimethylsilyl)amine with LiBun. J. Chem. Soc. Dalton Trans. 2001, 2494–2501. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Giesbrecht, G.R.; Rettig, S.J. Synthesis, Characterization, and Solution Dynamics of Alkali-Metal Chloride, Aluminate, and Borate Adducts of the Tridentate Amido Diphosphine Ligand Precursor LiN(SiMe2CH2PPri2)2. Organometallics 1997, 16, 725–736. [Google Scholar] [CrossRef]
- Johnson, C.S. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Koehne, I.; Gerstel, M.; Bruhn, C.; Reithmaier, J.P.; Benyoucef, M.; Pietschnig, R. Azido-Functionalized Aromatic Phosphonate Esters in RPOSS-Cage-Supported Lanthanide Ion (Ln = La, Nd, Dy, Er) Coordination. Inorg. Chem. 2021, 60, 5297–5309. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, E.J.; Berger, S.; Bräuer, P.; Kärger, J. High-Resolution DOSY NMR with Spins in Different Chemical Surroundings: Influence of Particle Exchange. J. Magn. Reson. 2002, 157, 124–131. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1·C6H14 1 | 2 (mol1) 2 | 2 (mol2) 3 | |||
|---|---|---|---|---|---|
| Li1 | Li2 2.578(4) Li3 3.359(4) O1 1.951(3) N1 2.032(3) N4 1.983(3) | Li1 | Li2 2.361(6) Li3 3.216(6) O1 1.913(4) N1 2.041(5) N6 2.020(4) | Li7 | Li8 2.353(6) Li9 3.103(6) O5 1.901(4) N11 2.055(4) N16 2.037(4) |
| Li2 | Li3 2.729(4) O2A 1.896(10) O2B 1.981(12) N1 2.112(3) N2 2.150(3) N5 2.065(3) | Li2 | Li3 2.915(6) O2 1.920(4) N1 2.042(4) N6 2.097(4) N7 2.335(5) | Li8 | Li9 3.002(6) O6A 1.908(13) O6B 1.95(3) N11 2.022(4) N16 2.104(4) N17 2.453(5) |
| Li3 | Li3I 2.595(5) O3 1.967(3) N2 2.407(3) N3 2.015(3) N5 2.084(3) N6 2.179(3) | Li3 | Li4 2.441(5) N2 1.965(4) N3 2.107(4) N7 1.992(4) N8 2.101(4) | Li9 | Li10 2.424(5) N12 1.985(4) N13 2.110(4) N17 1.985(4) N18 2.106(4) |
| Li4 | Li5 2.970(6) Li6 3.099(6) N3 2.086(4) N4 1.976(4) N8 2.107(4) N9 1.970(4) | Li10 | Li11 3.077(6) Li12 2.975(6) N13 2.092(4) N14 1.977(4) N18 2.094(4) N19 1.975(4) | ||
| Li5 | Li6 2.371(6) O3 1.906(4) N5 2.033(4) N9 2.365(5) N10 2.077(4) | Li11 | Li12 2.370(6) O7 1.917(4) N15 2.033(4) N20 2.074(5) | ||
| Li6 | O4 1.913(4) N5 2.066(5) N10 2.029(5) | Li12 | O8A 1.924(12) O8B 1.93(2) N14 2.375(5) N15 2.093(4) N20 2.024(4) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raza, A.; Mucci, A.; Nicolini, A.; Cornia, A. Structural Diversity of Lithium Oligo-α-Pyridylamides. Chemistry 2022, 4, 520-534. https://doi.org/10.3390/chemistry4020037
Raza A, Mucci A, Nicolini A, Cornia A. Structural Diversity of Lithium Oligo-α-Pyridylamides. Chemistry. 2022; 4(2):520-534. https://doi.org/10.3390/chemistry4020037
Chicago/Turabian StyleRaza, Arsen, Adele Mucci, Alessio Nicolini, and Andrea Cornia. 2022. "Structural Diversity of Lithium Oligo-α-Pyridylamides" Chemistry 4, no. 2: 520-534. https://doi.org/10.3390/chemistry4020037
APA StyleRaza, A., Mucci, A., Nicolini, A., & Cornia, A. (2022). Structural Diversity of Lithium Oligo-α-Pyridylamides. Chemistry, 4(2), 520-534. https://doi.org/10.3390/chemistry4020037