

Steering the Metal Precursor Location in Pd/Zeotype Catalysts and Its Implications for Catalysis

 , , , ,
, , , ,
Abstract
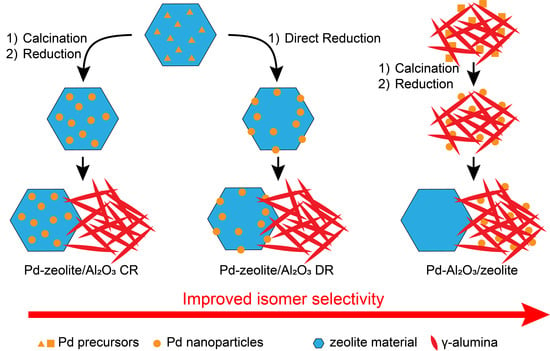
:
1. Introduction
2. Materials and Methods
2.1. Raw Materials
2.2. Catalyst Synthesis
2.2.1. Preparation of Catalysts with Pd on/in Zeotype Crystals
2.2.2. Preparation of Pd-On-Alumina Catalysts
2.3. Catalyst Characterization
2.3.1. Quantification of Metal and Acid Sites
2.3.2. Electron Microscopy and Energy Dispersive X-ray Spectroscopy
2.3.3. X-ray Photoelectron Spectroscopy
2.3.4. Nitrogen Physisorption
2.3.5. X-ray Diffraction
2.4. Hydroconversion of n-Heptane and n-Hexadecane
2.5. Calculation of Apparent Activation Energies
3. Results and Discussion
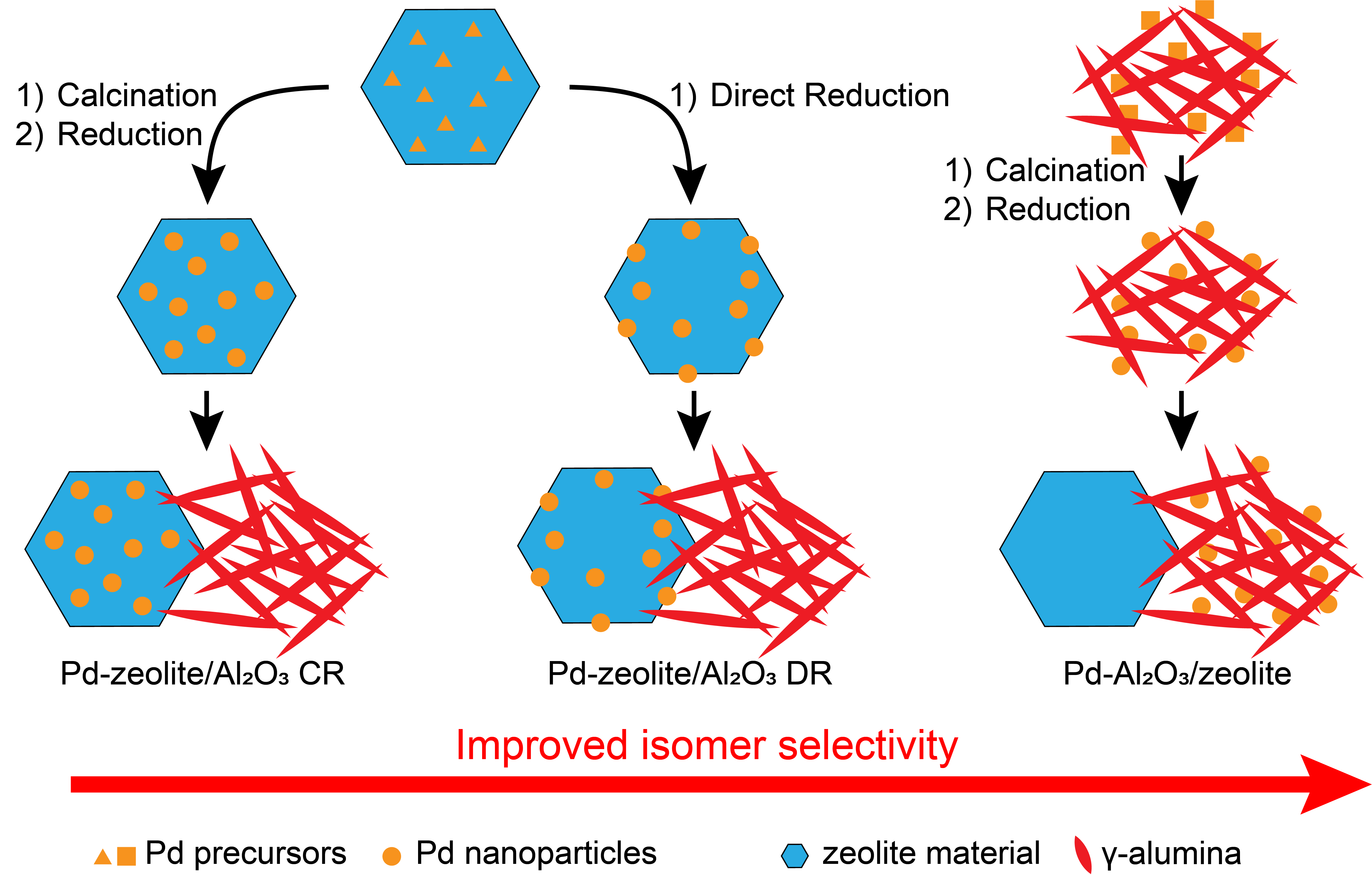
3.1. Acidity and Pd Weight Loading of Pd/SAPO-11/γ-Al2O3 and Pd/ZSM-22/γ-Al2O3 Catalysts
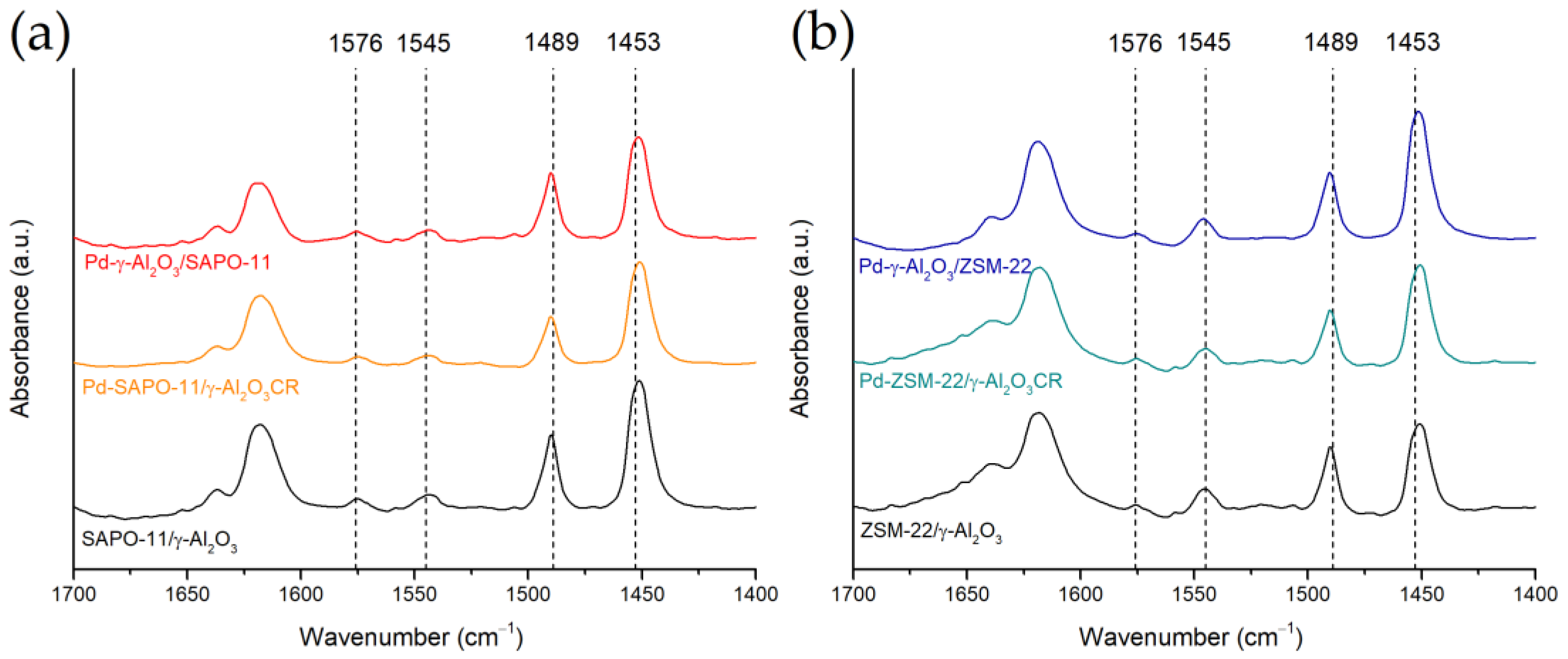
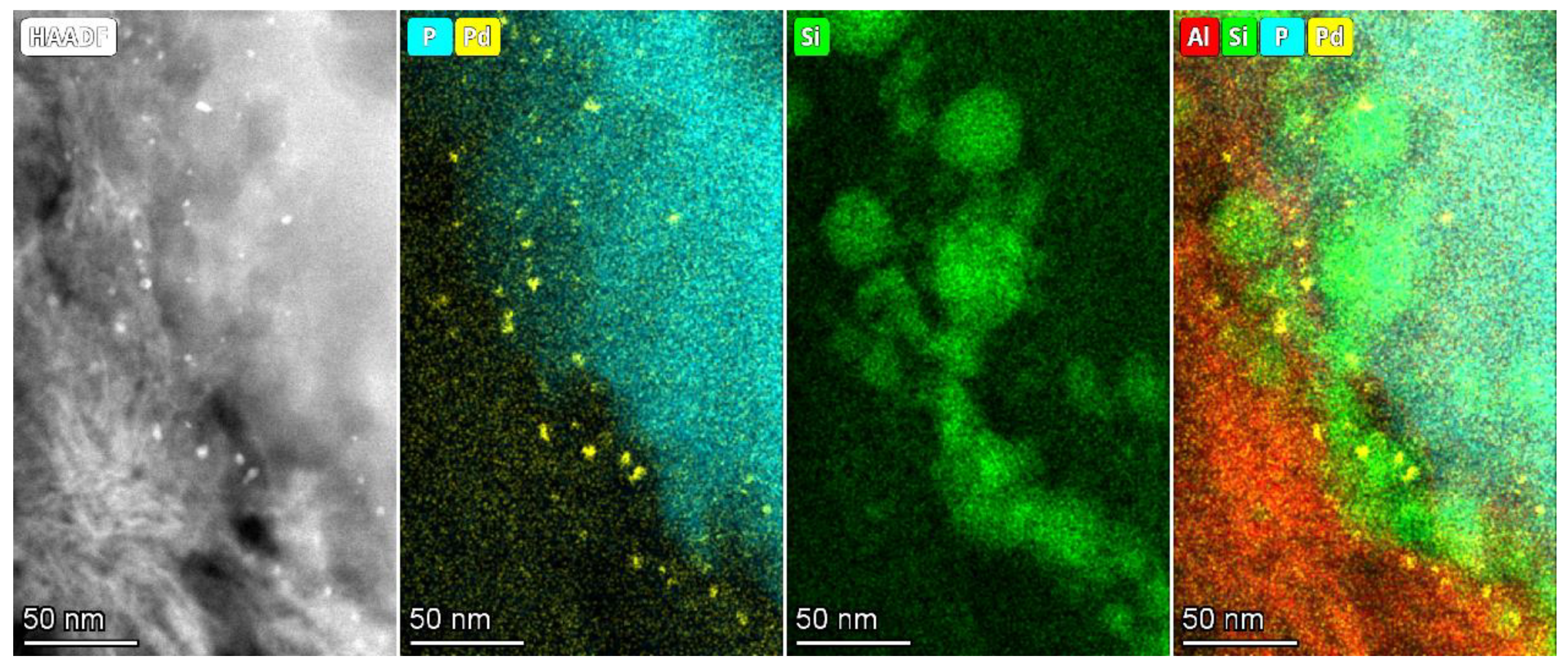
3.2. Determination of Pd Nanoparticle Location
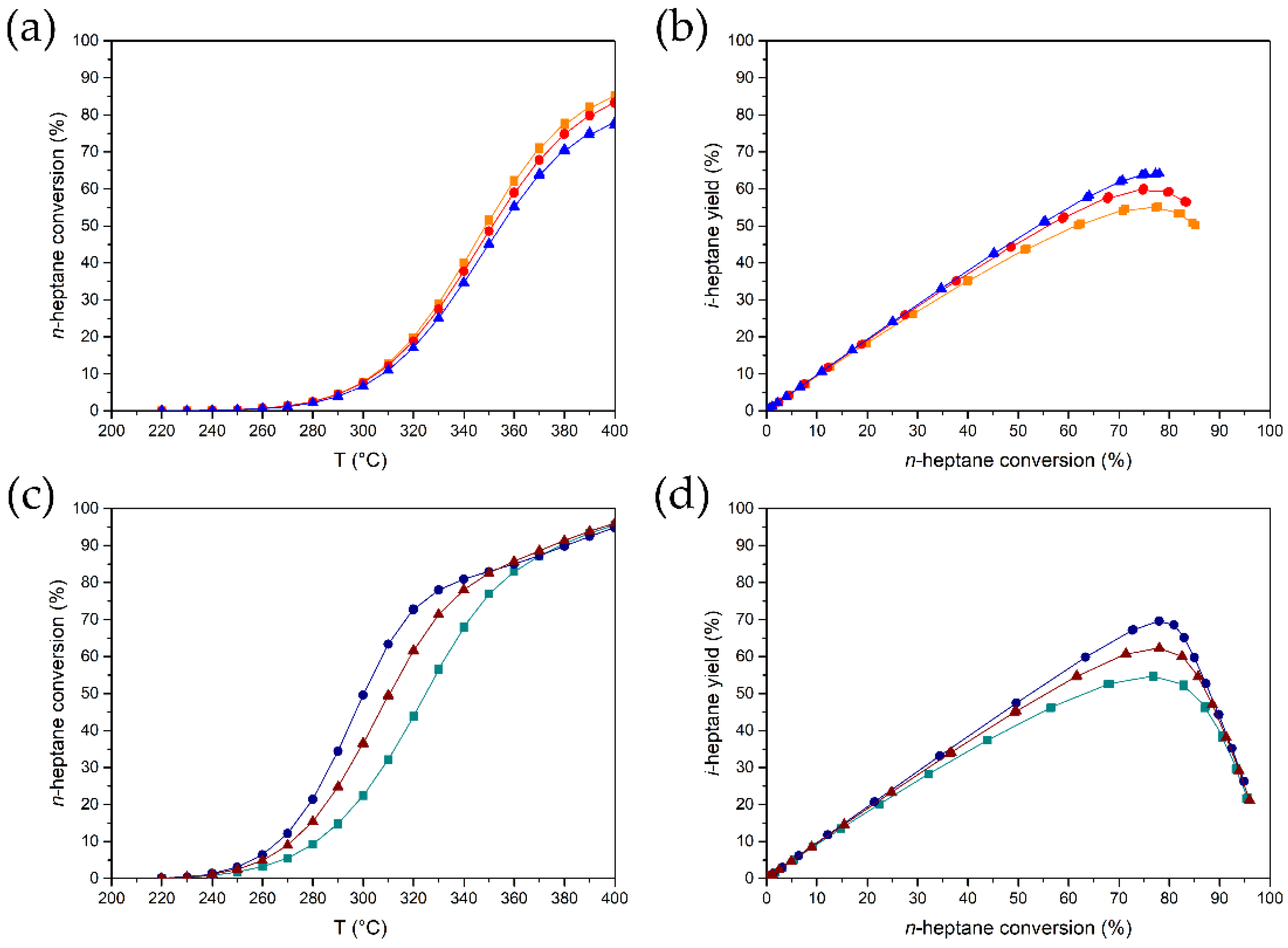
3.3. The Effect of the Metal Nanoparticle Location on n-Heptane Conversion
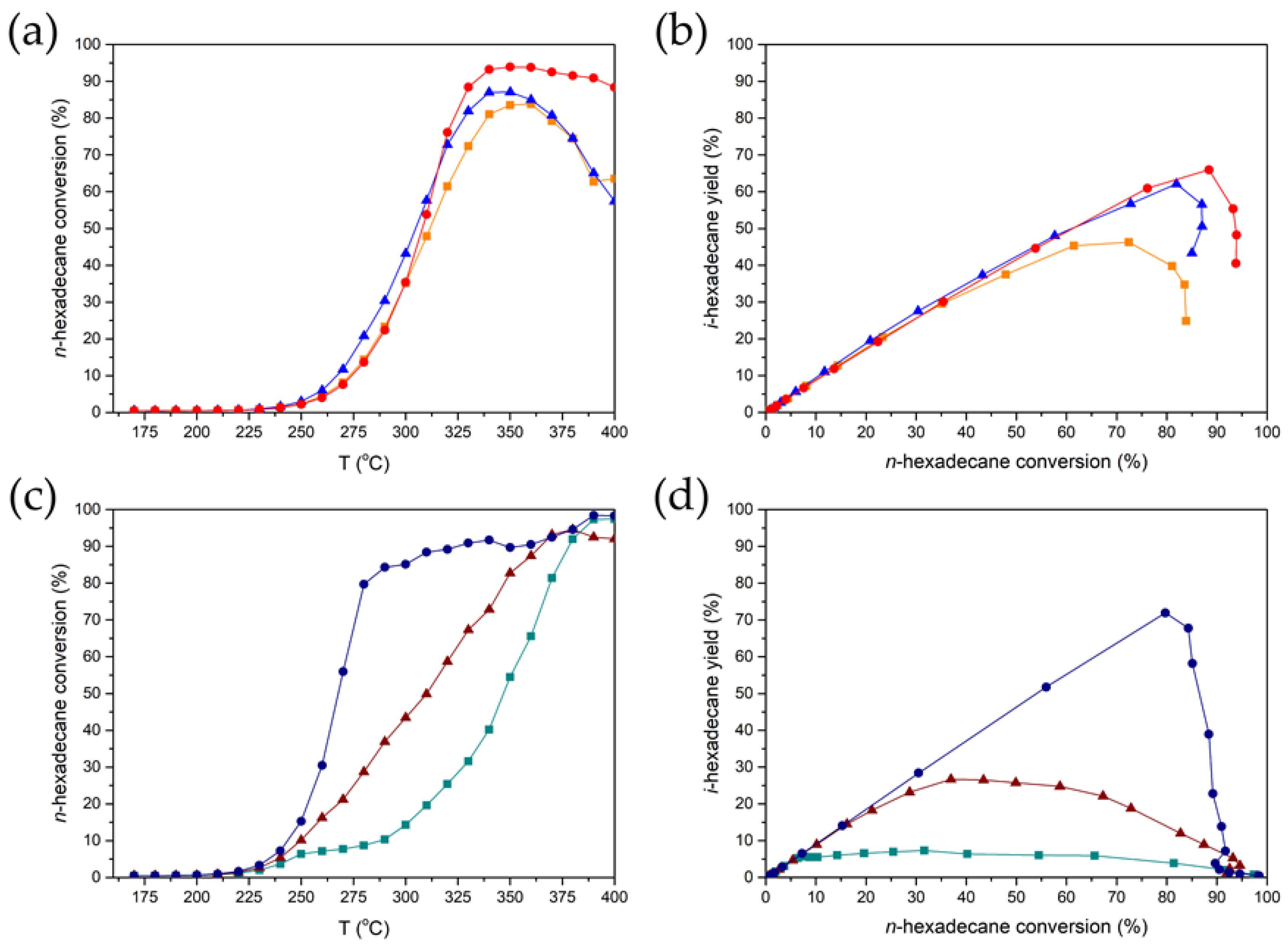
3.4. The Effect of the Metal Nanoparticle Location on n-Hexadecane Conversion
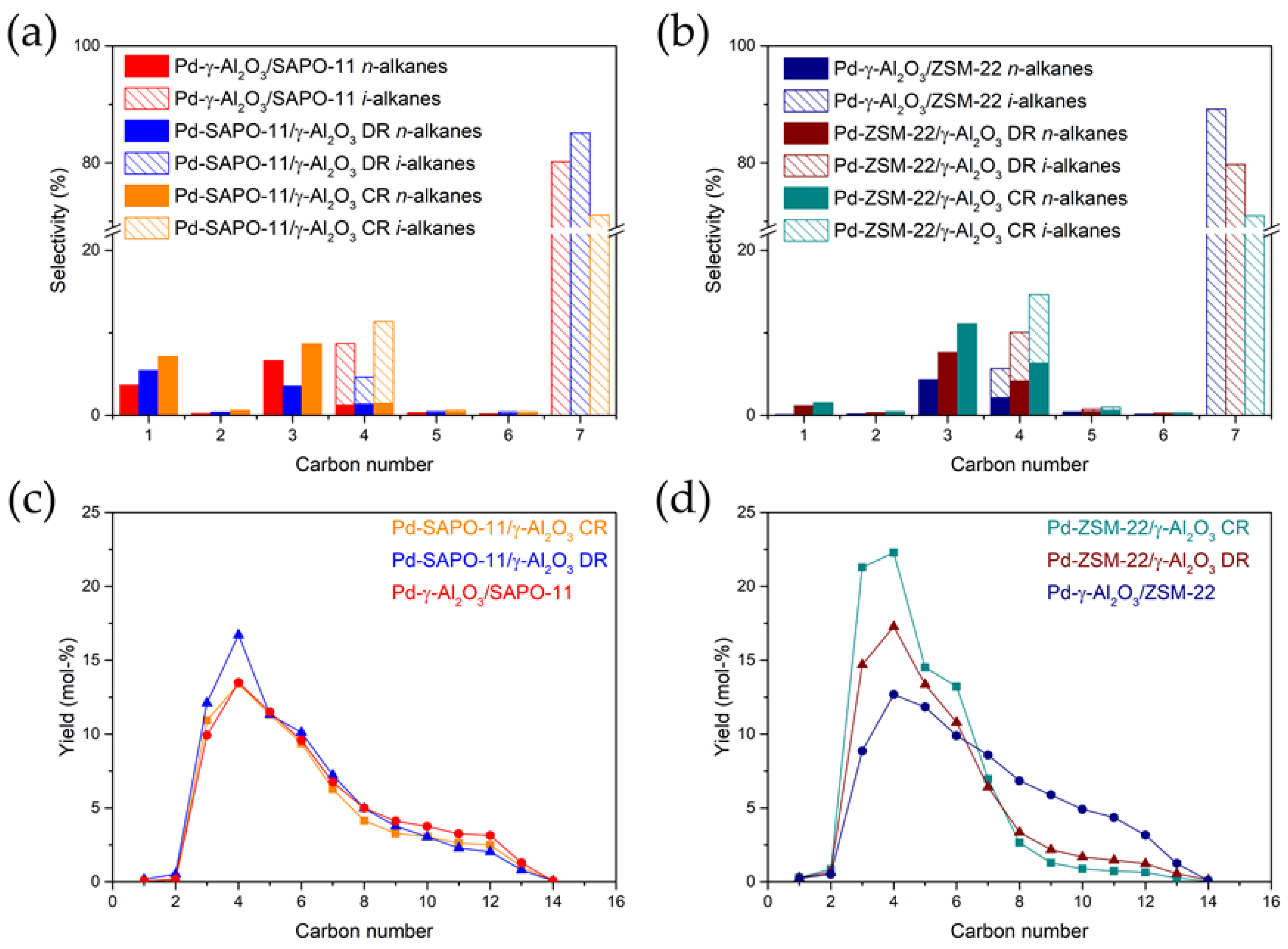
3.5. The Effect of the Metal Nanoparticle Location on Hydrocracking Selectivities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corma, A.; Martinez, A.; Martinezsoria, V.; Monton, J.B. Hydrocracking of Vacuum Gasoil on the Novel Mesoporous MCM-41 Aluminosilicate Catalyst. J. Catal. 1995, 153, 25–31. [Google Scholar] [CrossRef]
- Sastre, G.; Chica, A.; Corma, A. On the Mechanism of Alkane Isomerisation (Isodewaxing) with Unidirectional 10-Member Ring Zeolites. A Molecular Dynamics and Catalytic Study. J. Catal. 2000, 195, 227–236. [Google Scholar] [CrossRef]
- Chica, A.; Diaz, U.; Fornés, V.; Corma, A. Changing the Hydroisomerization to Hydrocracking Ratio of Long Chain Alkanes by Varying the Level of Delamination in Zeolitic (ITQ-6) Materials. Catal. Today 2009, 147, 179–185. [Google Scholar] [CrossRef]
- Martínez, C.; Corma, A. Inorganic Molecular Sieves: Preparation, Modification and Industrial Application in Catalytic Processes. Coord. Chem. Rev. 2011, 255, 1558–1580. [Google Scholar] [CrossRef] [Green Version]
- Mäki-Arvela, P.; Khel, T.A.K.; Azkaar, M.; Engblom, S.; Murzin, D.Y. Catalytic Hydroisomerization of Long-Chain Hydrocarbons for the Production of Fuels. Catalysts 2018, 8, 534. [Google Scholar] [CrossRef] [Green Version]
- Weitkamp, J. Catalytic Hydrocracking-Mechanisms and Versatility of the Process. ChemCatChem 2012, 4, 292–306. [Google Scholar] [CrossRef]
- Bouchy, C.; Hastoy, G.; Guillon, E.; Martens, J.A. Fischer-Tropsch Waxes Upgrading via Hydrocracking and Selective Hydroisomerization. Oil Gas Sci. Technol. 2009, 64, 91–112. [Google Scholar] [CrossRef] [Green Version]
- Sousa-Aguiar, E.F.; Noronha, F.B.; Faro, A. The Main Catalytic Challenges in GTL (Gas-to-Liquids) Processes. Catal. Sci. Technol. 2011, 1, 698–713. [Google Scholar] [CrossRef]
- Tran, N.H.; Bartlett, J.R.; Kannangara, G.S.K.; Milev, A.S.; Volk, H.; Wilson, M.A. Catalytic Upgrading of Biorefinery Oil from Micro-Algae. Fuel 2010, 89, 265–274. [Google Scholar] [CrossRef]
- Peng, B.; Yao, Y.; Zhao, C.; Lercher, J.A. Towards Quantitative Conversion of Microalgae Oil to Diesel-Range Alkanes with Bifunctional Catalysts. Angew. Chem. Int. Ed. 2012, 51, 2072–2075. [Google Scholar] [CrossRef]
- Kots, P.A.; Vance, B.C.; Vlachos, D.G. Polyolefin Plastic Waste Hydroconversion to Fuels, Lubricants, and Waxes: A Comparative Study. React. Chem. Eng. 2022, 7, 41–54. [Google Scholar] [CrossRef]
- Corma, A. Transformation of Hydrocarbons on Zeolite Catalysts. Catal. Letters 1993, 22, 33–52. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Whiting, G.T.; Dutta Chowdhury, A.; Weckhuysen, B.M. Zeolites and Zeotypes for Oil and Gas Conversion. Adv. Catal. 2015, 58, 143–314. [Google Scholar] [CrossRef]
- Wang, W.; Liu, C.-J.; Wu, W. Bifunctional Catalysts for the Hydroisomerization of N-Alkanes: The Effects of Metal–Acid Balance and Textural Structure. Catal. Sci. Technol. 2019, 9, 4162–4187. [Google Scholar] [CrossRef]
- Del Campo, P.; Martínez, C.; Corma, A. Activation and Conversion of Alkanes in the Confined Space of Zeolite-Type Materials. Chem. Soc. Rev. 2021, 50, 8511–8595. [Google Scholar] [CrossRef] [PubMed]
- Oenema, J.; Harmel, J.; Vélez, R.P.; Meijerink, M.J.; Eijsvogel, W.; Poursaeidesfahani, A.; Vlugt, T.J.H.; Zečević, J.; de Jong, K.P. Influence of Nanoscale Intimacy and Zeolite Micropore Size on the Performance of Bifunctional Catalysts for N-Heptane Hydroisomerization. ACS Catal. 2020, 10, 14245–14257. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P.S.F.; Silva, J.M.; Ribeiro, M.F.; Daudin, A.; Bouchy, C. Bifunctional Intimacy and Its Interplay with Metal-Acid Balance in Shaped Hydroisomerization Catalysts. ChemCatChem 2020, 12, 4582–4592. [Google Scholar] [CrossRef]
- Weisz, P.B. Stepwise Reaction via Intermediates on Separate Catalytic Centers. Science 1956, 123, 887–888. [Google Scholar] [CrossRef]
- Weisz, P.B.; Swegler, E.W. Stepwise Reaction on Separate Catalytic Centers: Isomerization of Saturated Hydrocarbons. Science 1957, 126, 31–32. [Google Scholar] [CrossRef]
- Zecevic, J.; Vanbutsele, G.; de Jong, K.P.; Martens, J.A. Nanoscale Intimacy in Bifunctional Catalysts for Selective Conversion of Hydrocarbons. Nature 2015, 528, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; van der Wal, L.I.; Yoshida, H.; Oenema, J.; Harmel, J.; Zhang, Z.; Sunley, G.; Zečević, J.; de Jong, K.P. Impact of the Spatial Organization of Bifunctional Metal–Zeolite Catalysts on the Hydroisomerization of Light Alkanes. Angew. Chem. Int. Ed. 2020, 59, 3592–3600. [Google Scholar] [CrossRef]
- Cheng, K.; Smulders, L.C.J.; van der Wal, L.I.; Oenema, J.; Meeldijk, J.D.; Visser, N.L.; Sunley, G.; Roberts, T.; Xu, Z.; Doskocil, E.; et al. Maximizing Noble Metal Utilization in Solid Catalysts by Control of Nanoparticle Location. Science 2022, 377, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, W.; Seo, Y.; Kim, J.C.; Ryoo, R. N-Heptane Hydroisomerization over Pt/MFI Zeolite Nanosheets: Effects of Zeolite Crystal Thickness and Platinum Location. J. Catal. 2013, 301, 187–197. [Google Scholar] [CrossRef]
- Noh, G.; Shi, Z.; Zones, S.I.; Iglesia, E. Isomerization and Β-Scission Reactions of Alkanes on Bifunctional Metal-Acid Catalysts: Consequences of Confinement and Diffusional Constraints on Reactivity and Selectivity. J. Catal. 2018, 368, 389–410. [Google Scholar] [CrossRef]
- Noh, G.; Zones, S.I.; Iglesia, E. Isomer Sieving and the Selective Formation of Terminal Methyl Isomers in Reactions of Linear Alkanes on One-Dimensional Zeolites. J. Catal. 2019, 377, 255–270. [Google Scholar] [CrossRef]
- Oenema, J.; Hofmann, J.P.; Hensen, E.J.M.; Zečević, J.; de Jong, K.P. Assessment of the Location of Pt Nanoparticles in Pt/Zeolite Y/Γ-Al2O3 Composite Catalysts. ChemCatChem 2020, 12, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höchtl, M.; Jentys, A.; Vinek, H. Hydroisomerization of Heptane Isomers over Pd/SAPO Molecular Sieves: Influence of the Acid and Metal Site Concentration and the Transport Properties on the Activity and Selectivity. J. Catal. 2000, 190, 419–432. [Google Scholar] [CrossRef]
- Höchtl, M.; Jentys, A.; Vinek, H. Alkane Conversion over Pd/SAPO Molecular Sieves: Influence of Acidity, Metal Concentration and Structure. Catal. Today 2001, 65, 171–177. [Google Scholar] [CrossRef]
- Wang, D.; Liu, J.; Cheng, X.; Kang, X.; Wu, A.; Tian, C.; Fu, H. Trace Pt Clusters Dispersed on SAPO-11 Promoting the Synergy of Metal Sites with Acid Sites for High-Effective Hydroisomerization of N-Alkanes. Small Methods 2019, 3, 1800510. [Google Scholar] [CrossRef]
- Sinha, A.K.; Sivasanker, S. Hydroisomerization of N-Hexane over Pt-SAPO-11 and Pt-SAPO-31 Molecular Sieves. Catal. Today 1999, 49, 293–302. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, Z.; Teng, F.; Wen, G.; Xu, Y.; Xu, Z.; Lin, L. Hydroisomerization of Long-Chain Alkane over Pt/SAPO-11 Catalysts Synthesized from Nonaqueous Media. Catal. Lett. 2005, 103, 109–116. [Google Scholar] [CrossRef]
- Van den Broek, A.C.M.; van Grondelle, J.; van Santen, R.A. Preparation of Highly Dispersed Platinum Particles in HZSM-5 Zeolite: A Study of the Pretreatment Process of [Pt(NH3)4]2+. J. Catal. 1997, 167, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Reagan, W.J.; Chester, A.W.; Kerr, G.T. Studies of the Thermal Decomposition and Catalytic Properties of Some Platinum and Palladium Ammine Zeolites. J. Catal. 1981, 69, 89–100. [Google Scholar] [CrossRef]
- De Graaf, J.; van Dillen, A.J.; de Jong, K.P.; Koningsberger, D.C. Preparation of Highly Dispersed Pt Particles in Zeolite Y with a Narrow Particle Size Distribution: Characterization by Hydrogen Chemisorption, TEM, EXAFS Spectroscopy, and Particle Modeling. J. Catal. 2001, 203, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, W.; Zhang, M.; Wang, W.; Zhang, X.-H.; Lu, F.; Cheng, K.; Zhang, Q.; Wang, Y. Boosting Propane Dehydroaromatization by Confining PtZn Alloy Nanoparticles within H-ZSM-5 Crystals. Catal. Sci. Technol. 2022, 10, 4513–4521. [Google Scholar] [CrossRef]
- Goel, S.; Wu, Z.; Zones, S.I.; Iglesia, E. Synthesis and Catalytic Properties of Metal Clusters Encapsulated within Small-Pore (SOD, GIS, ANA) Zeolites. J. Am. Chem. Soc. 2012, 134, 17688–17695. [Google Scholar] [CrossRef]
- van der Wal, L.I.; Oenema, J.; Smulders, L.C.J.; Samplonius, N.J.; Nandpersad, K.R.; Zečević, J.; de Jong, K.P. Control and Impact of Metal Loading Heterogeneities at the Nanoscale on the Performance of Pt/Zeolite Y Catalysts for Alkane Hydroconversion. ACS Catal. 2021, 11, 3842–3855. [Google Scholar] [CrossRef]
- Winter, F.; Bezemer, G.L.; van der Spek, C.; Meeldijk, J.D.; van Dillen, A.J.; Geus, J.W.; de Jong, K.P. TEM and XPS Studies to Reveal the Presence of Cobalt and Palladium Particles in the Inner Core of Carbon Nanofibers. Carbon 2005, 43, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Sun, Q.; Bai, R.; Li, X.; Guo, G.; Yu, J. In Situ Confinement of Ultrasmall Pd Clusters within Nanosized Silicalite-1 Zeolite for Highly Efficient Catalysis of Hydrogen Generation. J. Am. Chem. Soc. 2016, 138, 7484–7487. [Google Scholar] [CrossRef]
- Cho, H.R.; Regalbuto, J.R. The Rational Synthesis of Pt-Pd Bimetallic Catalysts by Electrostatic Adsorption. Catal. Today 2015, 246, 143–153. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Busca, G. Spectroscopic Characterization of the Acid Properties of Metal Oxide Catalysts. Catal. Today 1998, 41, 191–206. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Nab, S.; Weckhuysen, B.M. Probing Acid Sites in Solid Catalysts with Pyridine UV-Vis Spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 21647–21659. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, U.; Rigutto, M.S.; Zuidema, E.; Jansen, A.P.J.; Poursaeidesfahani, A.; Sharma, S.; Dubbeldam, D.; Vlugt, T.J.H. Kinetics of Zeolite-Catalyzed Heptane Hydroisomerization and Hydrocracking with CBMC-Modeled Adsorption Terms: Zeolite Beta as a Large Pore Base Case. J. Catal. 2022, 415, 37–50. [Google Scholar] [CrossRef]
- Guisnet, M.; Alvarez, F.; Giannetto, G.; Perot, G. Hydroisomerization and Hydrocracking of N-Heptane on PtH Zeolites. Effect of the Porosity and of the Distribution of Metallic and Acid Sites. Catal. Today 1987, 1, 415–433. [Google Scholar] [CrossRef]
- Alvarez, F.; Ribeiro, F.R.; Perot, G.; Thomazeau, C.; Guisnet, M. Hydroisomerization and Hydrocracking of Alkanes. J. Catal. 1996, 162, 179–189. [Google Scholar] [CrossRef]
- Bagshaw, S.A.; Cooney, R.P. FTIR Surface Site Analysis of Pillared Clays Using Pyridine Probe Species. Chem. Mater. 1993, 5, 1101–1109. [Google Scholar] [CrossRef]
- Barzetti, T.; Selli, E.; Moscotti, D.; Forni, L. Pyridine and Ammonia as Probes for FTIR Analysis of Solid Acid Catalysts. J. Chem. Soc. Faraday Trans. 1996, 92, 1401–1407. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Muñoz-Murillo, A.; Bouhmadi, A.; Cecius, M.; Diefenbach, S.; Weckhuysen, B.M. The Multifaceted Role of Methylaluminoxane in Metallocene-Based Olefin Polymerization Catalysis. Macromolecules 2018, 51, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Lok, B.M.; Messina, C.A.; Patton, R.L.; Gajek, R.T.; Cannan, T.R.; Flanigen, E.M. Silicoaluminophosphate Molecular Sieves: Another New Class of Microporous Crystalline Inorganic Solids. J. Am. Chem. Soc. 1984, 106, 6092–6093. [Google Scholar] [CrossRef]
- Martens, J.A.; Grobet, P.J.; Jacobs, P.A. Catalytic Activity and Si, Al, P Ordering in Microporous Silicoaluminophosphates of the SAPO-5, SAPO-11, and SAPO-37 Type. J. Catal. 1990, 126, 299–305. [Google Scholar] [CrossRef]
- Martens, J.A.; Tielen, M.; Jacobs, P.A. Attempts to Rationalize the Distribution of Hydrocracked Products. III. Mechanistic Aspects of Isomerization and Hydrocracking of Branched Alkanes on Ideal Bifunctional Large-Pore Zeolite Catalysts. Catal. Today 1987, 1, 435–453. [Google Scholar] [CrossRef]
- Gobin, O.C.; Reitmeier, S.J.; Jentys, A.; Lercher, J.A. Role of the Surface Modification on the Transport of Hexane Isomers in ZSM-5. J. Phys. Chem. C 2011, 115, 1171–1179. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Pd Weight Loading a (wt%) | Number of Strong Acid Sites b (mmol/g) | Number of Brønsted Acid Sites (mmol/g) c | Pd/(Si+Al) Surface d (at/at) | Pd/(Si+Al) Bulk a (at/at) |
|---|---|---|---|---|---|
| Pd-γ-Al2O3/SAPO-11 | 0.49 | 0.29 | 0.015 | 0.0037 ± 0.0001 | 0.0036 |
| Pd-SAPO-11/γ-Al2O3 DR | 0.45 | 0.26 | n.d. | 0.0020 ± 0.0001 | 0.0032 |
| Pd-SAPO-11/γ-Al2O3 CR | 0.41 | 0.25 | 0.018 | 0.0018 ± 0.0001 | 0.0028 |
| Pd-γ-Al2O3/ZSM-22 | 0.50 | 0.37 | 0.023 | 0.0028 ± 0.0001 | 0.0026 |
| Pd-ZSM-22/γ-Al2O3 DR | 0.44 | 0.35 | n.d. | 0.0014 ± 0.0002 | 0.0023 |
| Pd-ZSM-22/γ-Al2O3 CR | 0.41 | 0.33 | 0.019 | 0.0010 ± 0.0001 | 0.0022 |
| Catalyst | Ea n-Heptane Conversion (kJ mol−1) | Ea n-Hexadecane Conversión (kJ mol−1) |
|---|---|---|
| Pd-γ-Al2O3/SAPO-11 | 148 ± 1 | 143 ± 4 |
| Pd-SAPO-11/γ-Al2O3 DR | 152 ± 1 | 140 ± 2 |
| Pd-SAPO-11/γ-Al2O3 CR | 150 ± 1 | 140 ± 1 |
| Pd-γ-Al2O3/ZSM-22 | 173 ± 4 | 148 ± 8 |
| Pd-ZSM-22/γ-Al2O3 DR | 160 ± 4 | 119 ± 2 |
| Pd-ZSM-22/γ-Al2O3 CR | 135 ± 1 | 85 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smulders, L.C.J.; van de Minkelis, J.H.; Meeldijk, J.D.; Tang, M.; Liutkova, A.; Cheng, K.; Roberts, S.T.; Sunley, G.J.; Hensen, E.J.M.; de Jongh, P.E.; et al. Steering the Metal Precursor Location in Pd/Zeotype Catalysts and Its Implications for Catalysis. Chemistry 2023, 5, 348-364. https://doi.org/10.3390/chemistry5010026
Smulders LCJ, van de Minkelis JH, Meeldijk JD, Tang M, Liutkova A, Cheng K, Roberts ST, Sunley GJ, Hensen EJM, de Jongh PE, et al. Steering the Metal Precursor Location in Pd/Zeotype Catalysts and Its Implications for Catalysis. Chemistry. 2023; 5(1):348-364. https://doi.org/10.3390/chemistry5010026
Chicago/Turabian StyleSmulders, Luc C. J., Johan H. van de Minkelis, Johannes D. Meeldijk, Min Tang, Anna Liutkova, Kang Cheng, S. Tegan Roberts, Glenn J. Sunley, Emiel J. M. Hensen, Petra E. de Jongh, and et al. 2023. "Steering the Metal Precursor Location in Pd/Zeotype Catalysts and Its Implications for Catalysis" Chemistry 5, no. 1: 348-364. https://doi.org/10.3390/chemistry5010026
APA StyleSmulders, L. C. J., van de Minkelis, J. H., Meeldijk, J. D., Tang, M., Liutkova, A., Cheng, K., Roberts, S. T., Sunley, G. J., Hensen, E. J. M., de Jongh, P. E., & de Jong, K. P. (2023). Steering the Metal Precursor Location in Pd/Zeotype Catalysts and Its Implications for Catalysis. Chemistry, 5(1), 348-364. https://doi.org/10.3390/chemistry5010026