
Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles



Abstract
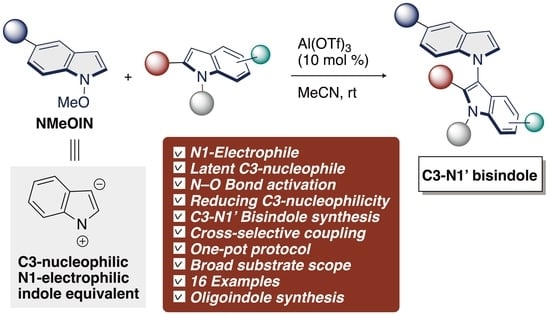
:
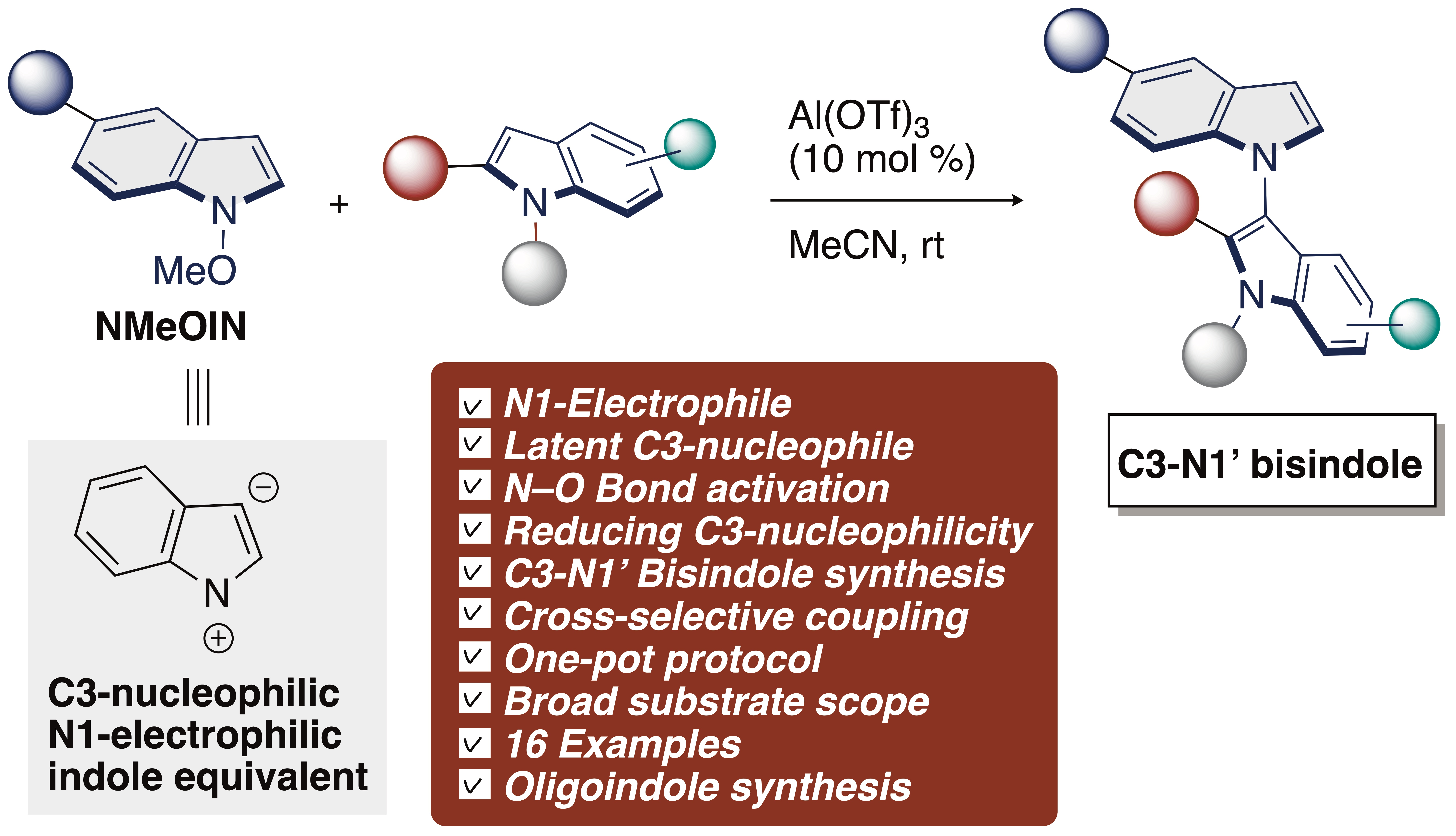
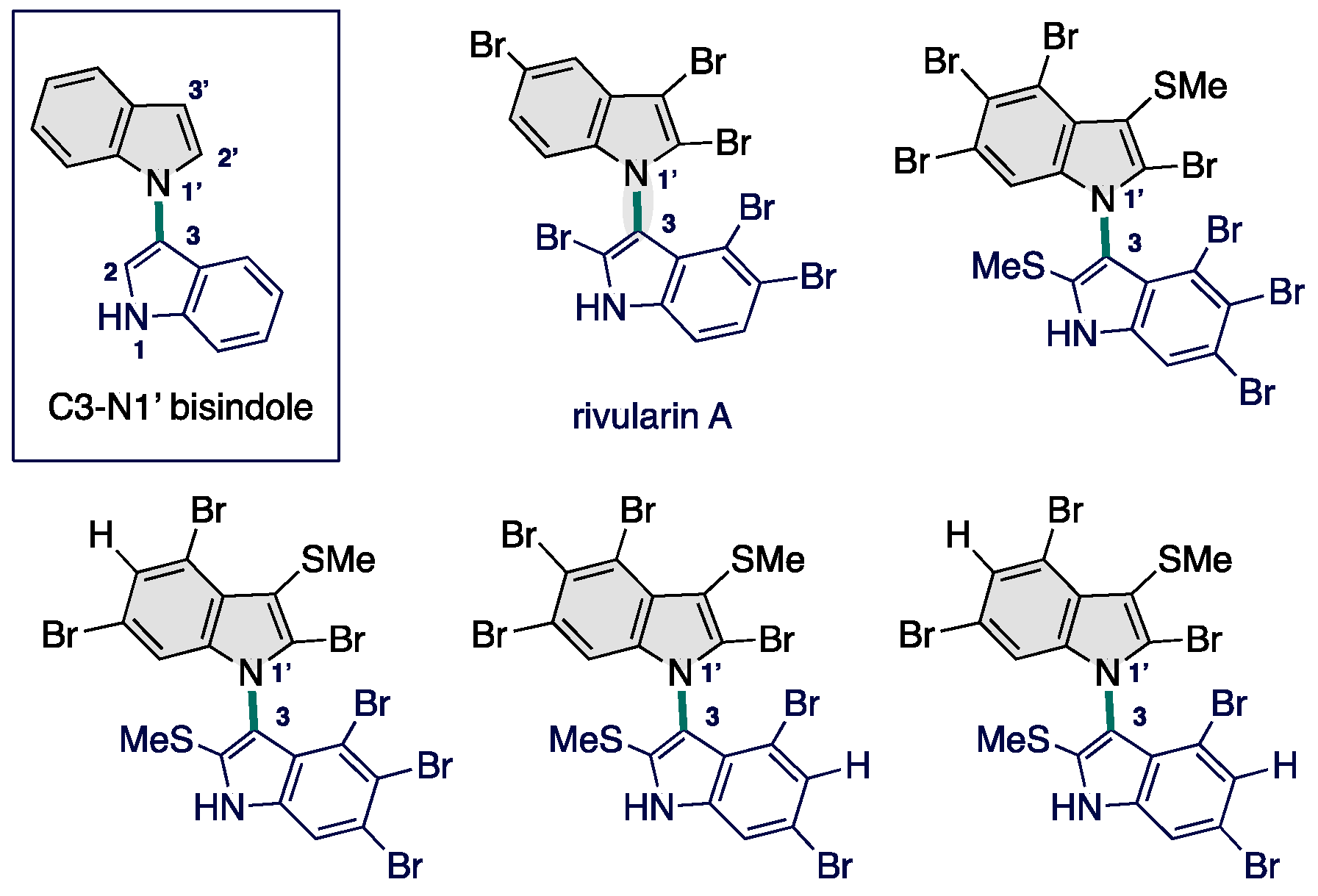
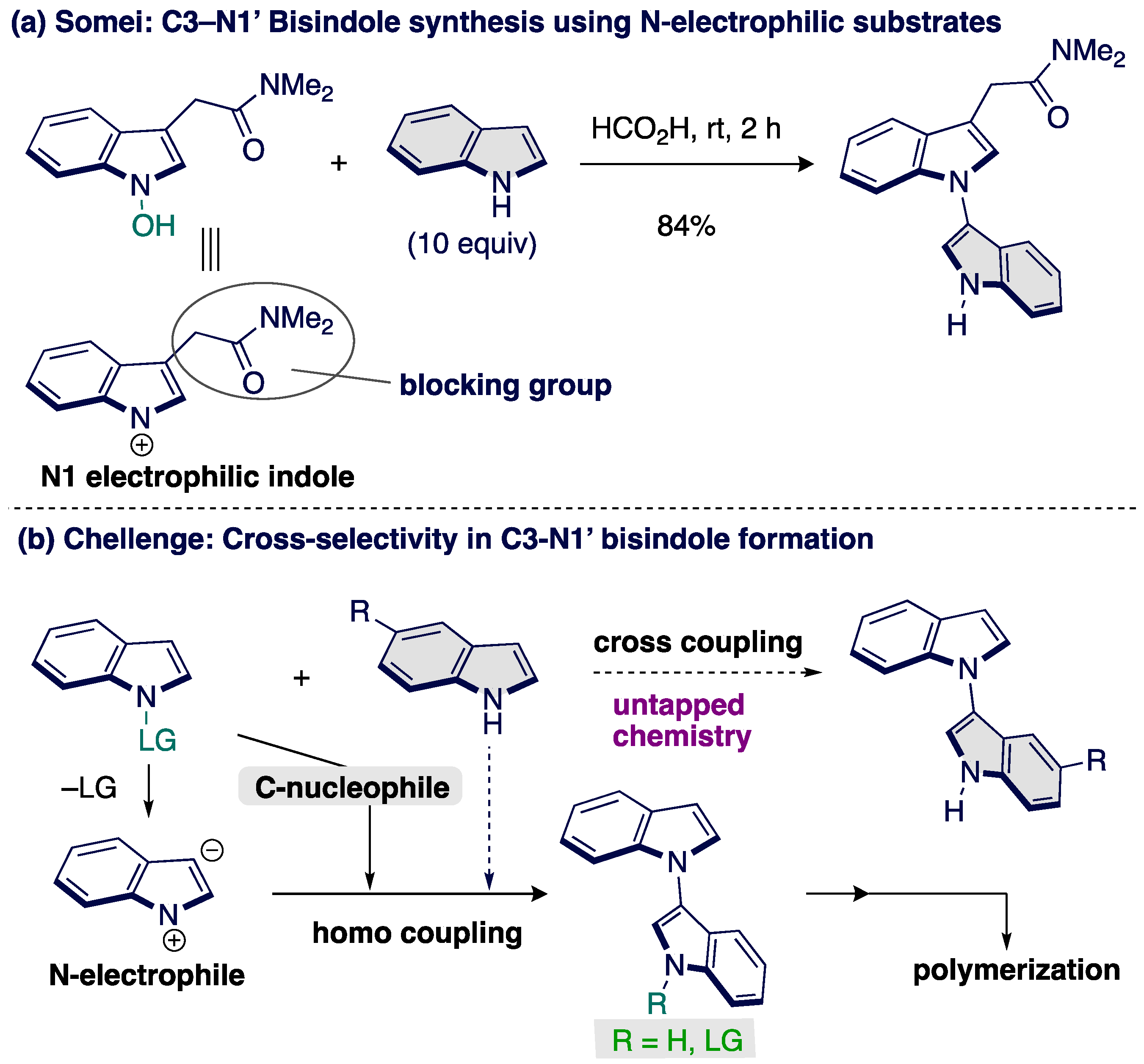
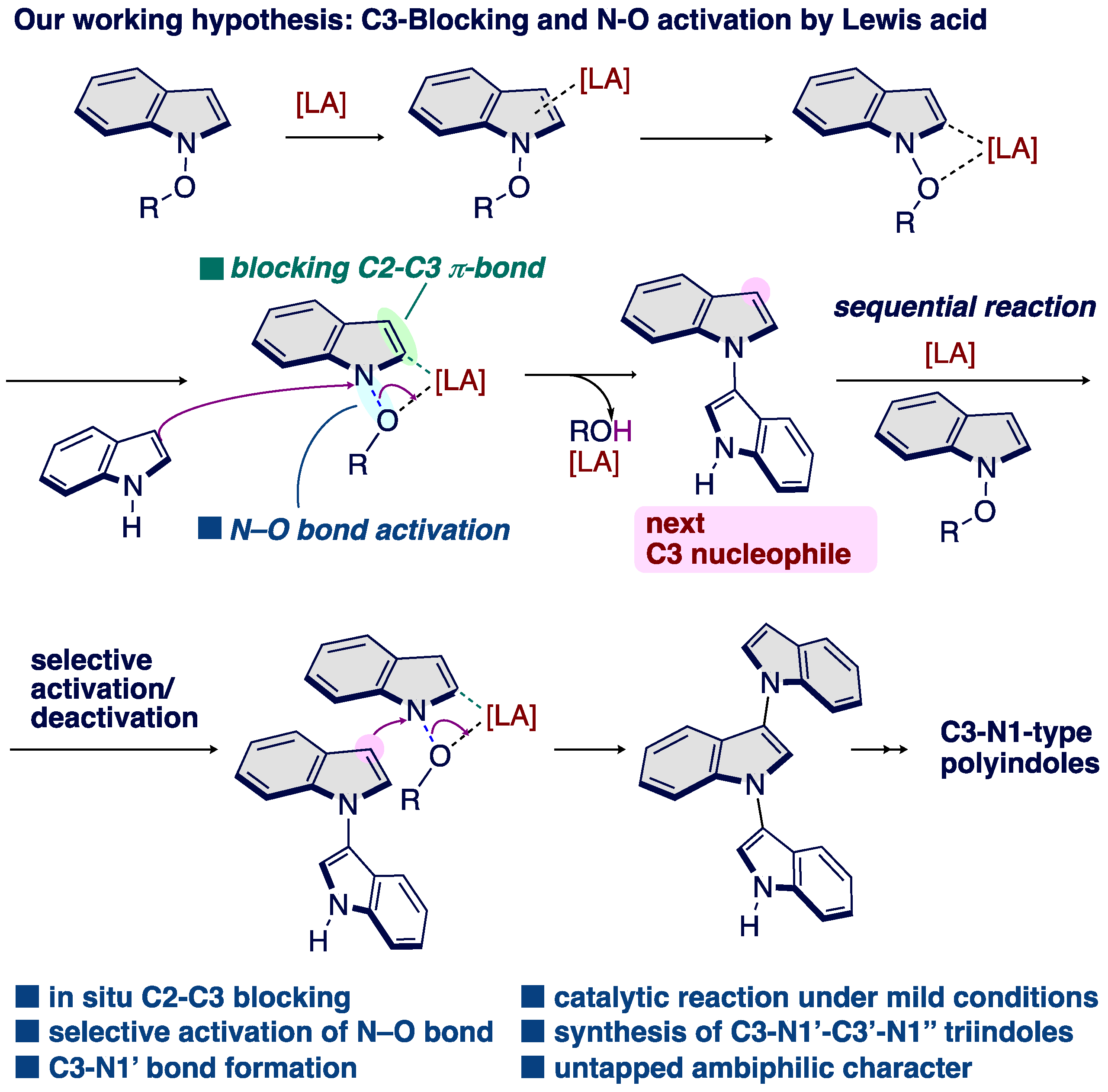
1. Introduction
2. Materials and Methods
2.1. General Procedure for Synthesis of NMeOINs [54,55]
2.2. General Procedure for Synthesis of 1′H-1,3′-Biindole Derivatives (Scheme 3)
2.3. General Procedure for Synthesis of Oligoindoles (Scheme 4)
3. Results and Discussion
3.1. Optimization of Reaction Conditions
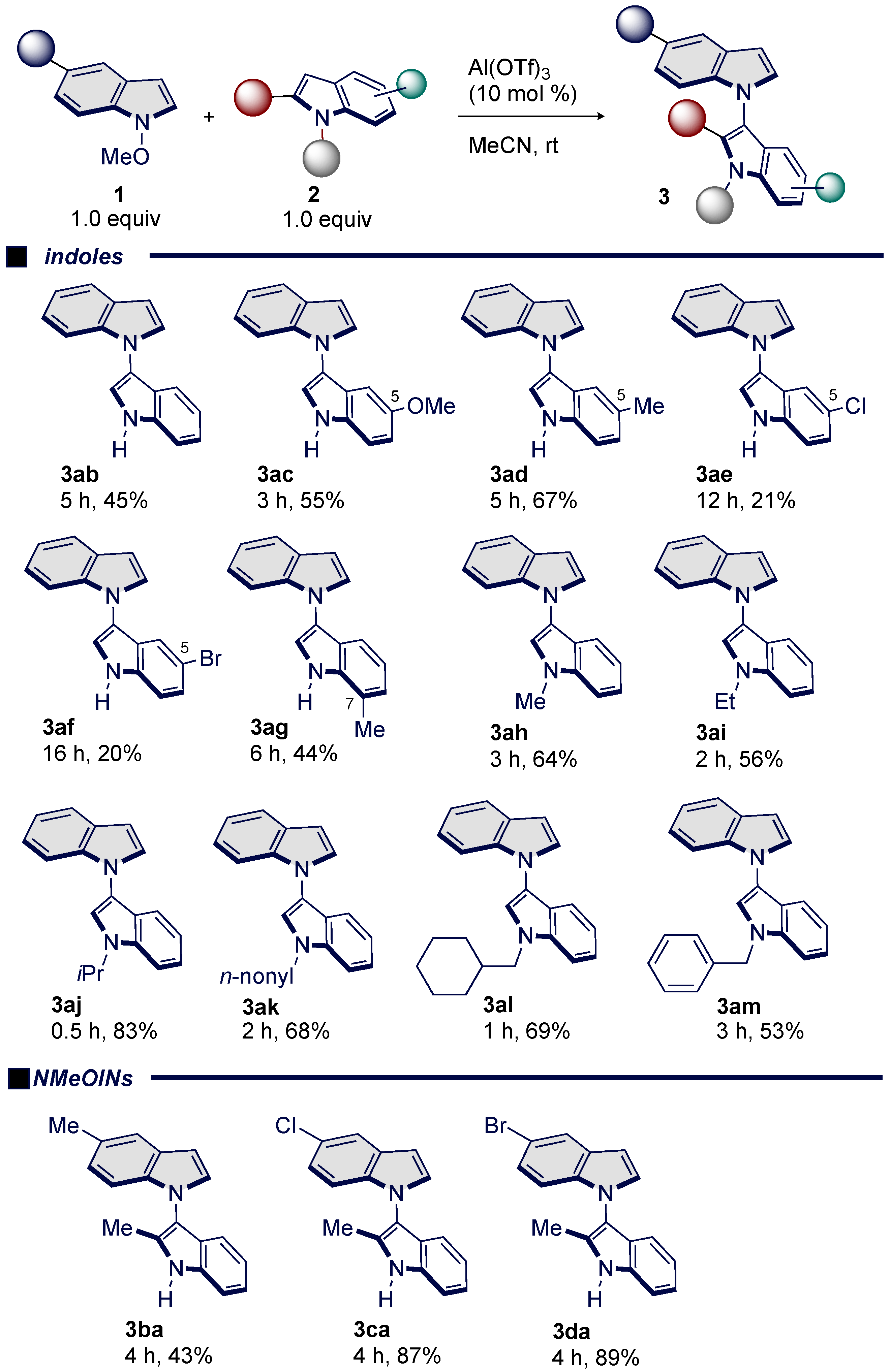
3.2. Scope and Limitations
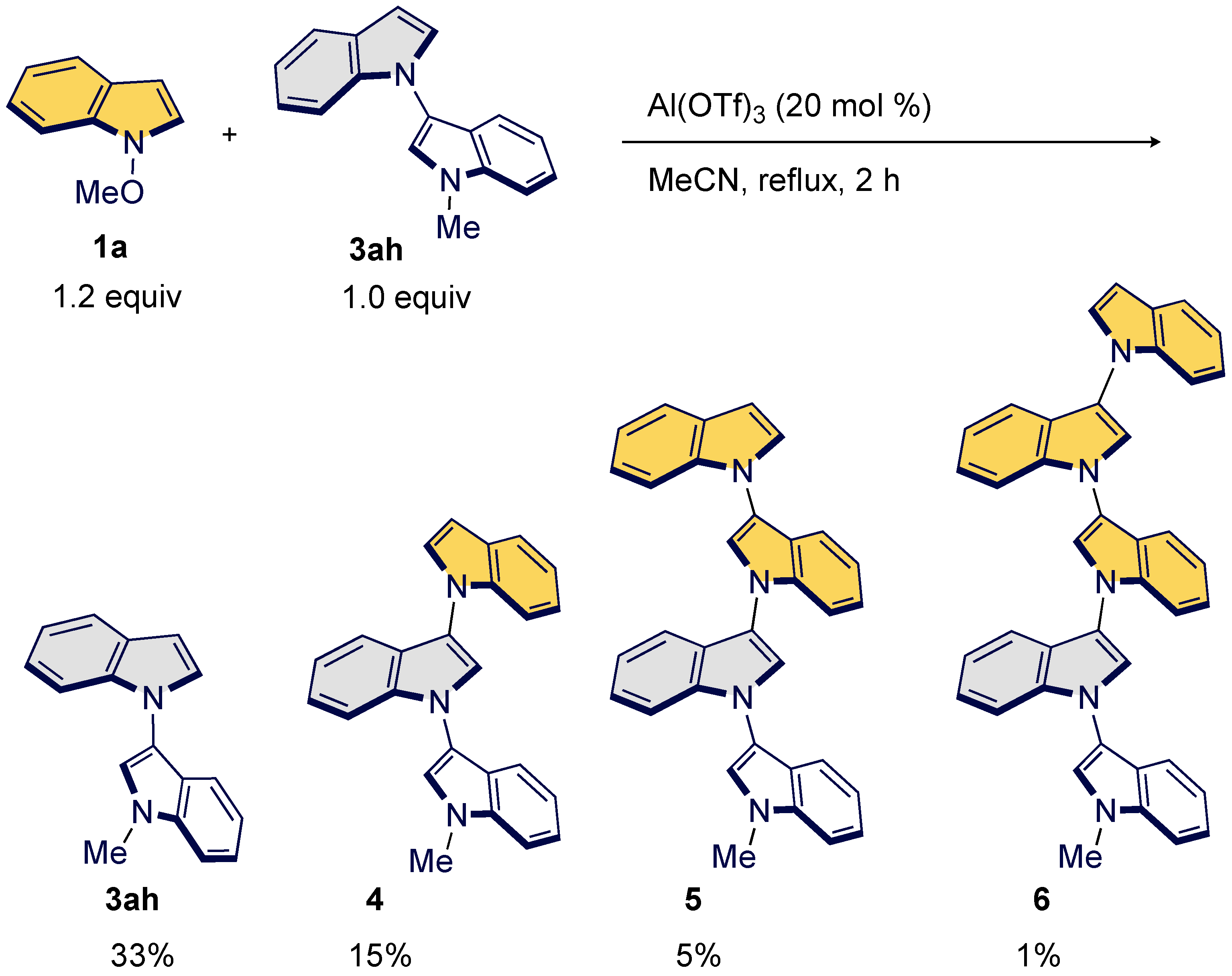
3.3. Synthesis of Oligoindoles
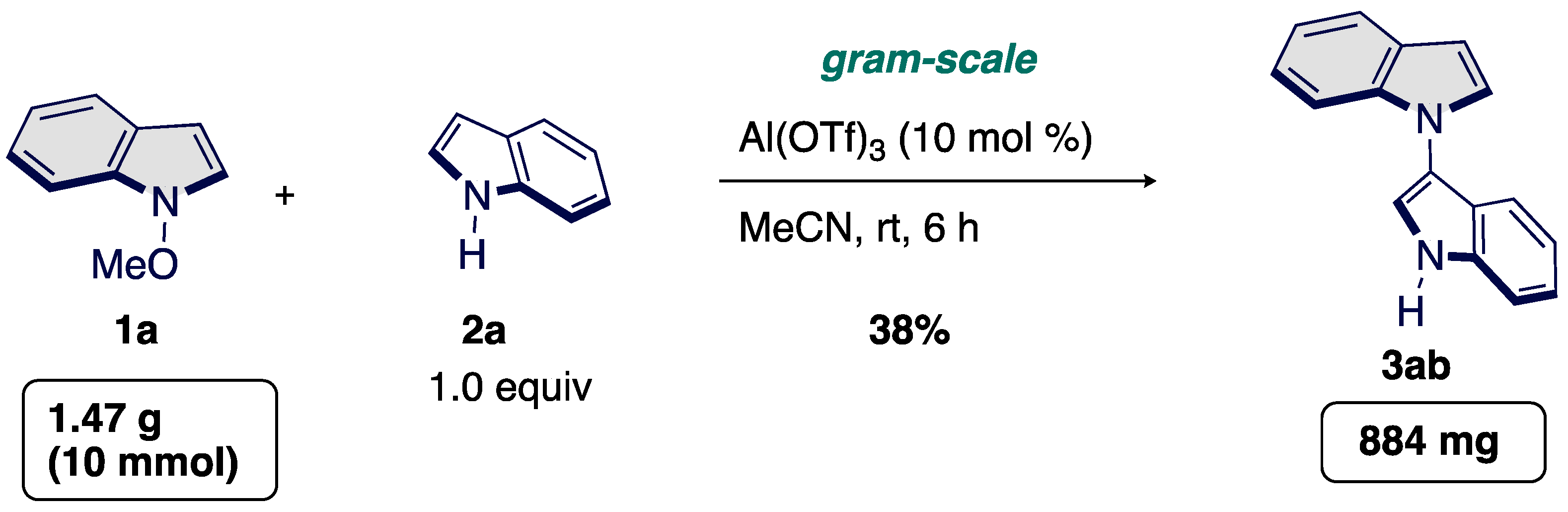
3.4. Scalability of the Aluminum-Catalyzed Cross Selective C3–N1′Cross-Coupling Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Espejo, V.R.; Rainier, J.D. An expeditious synthesis of C(3)-N(1′) heterodimeric indolines. J. Am. Chem. Soc. 2008, 130, 12894–12895. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S. Total synthesis of (±)-psychotriamine. J. Am. Chem. Soc. 2008, 130, 10886–10887. [Google Scholar] [CrossRef]
- Espejo, V.R.; Rainier, J.D. Total synthesis of kapakahine E and F. Org. Lett. 2010, 12, 2154–2157. [Google Scholar] [CrossRef]
- Pérez-Balado, C.; de Lera, Á.R. Concise total synthesis and structural revision of (+)-pestalazine B. Org. Biomol. Chem. 2010, 8, 5179–5186. [Google Scholar] [CrossRef]
- Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P.S. Total synthesis of guided structure elucidation of (+)-psychotetramine. Angew. Chem. Int. Ed. 2011, 50, 2716–2719. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xia, T.; Yao, L.; Deng, H.; Liao, X. Enantioselective and diastereoselective azo-coupling/iminium-cyclizations: A unified strategy for the total syntheses of (–)-psychotriasine and (+)-pestalazine B. Chem. Sci. 2015, 6, 3599–3605. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.A.; Chisholm, J.D. Lewis acid catalyzed displacement of trichloroacetamidates in the synthesis of functionalized pyrroloindolines. Org. Lett. 2016, 18, 4100–4103. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yi, J.-C.; Xheng, Z.-B.; Tang, Y.; Dai, L.-X.; You, S.-L. Enantioselective synthesis of 3a-amino-pyrroloindolines by copper-catalyzed direct asymmetric dearomative amination of tryptamines. Angew. Chem. Int. Ed. 2016, 55, 751–754. [Google Scholar] [CrossRef]
- Dai, J.; Xiong, D.; Yuan, T.; Liu, J.; Chen, T.; Shao, Z. Chiral primary amine catalysis for asymmetric Mannich reactions of aldehydes with ketimines: Stereoselectivity and reactivity. Angew. Chem. Int. Ed. 2017, 56, 12697–12701. [Google Scholar] [CrossRef]
- Nelson, B.M.; Loach, R.P.; Schiesser, S.; Movassaghi, M. Concise total synthesis of (+)-asperazine A and (+)-pestalazine B. Org. Biomol. Chem. 2018, 16, 202–207. [Google Scholar] [CrossRef]
- Gentry, E.C.; Rono, L.J.; Hale, M.E.; Matsuura, R.; Knowles, R.R. Enantioselective synthesis of pyrroloindolines via noncovalent stabilization of indole radical cations to the synthesis of alkaloid natural products. J. Am. Chem. Soc. 2018, 140, 3394–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakamata, H.; Ueda, H.; Tokuyama, H. Construction of indole structure on pyrroloindolines via AgNTf2-mediated amination/cyclization cascade: Application to total synthesis of (+)-pestazine B. Org. Lett. 2019, 21, 4205–4209. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Wells, R.J. A series of chiral polybrominated biindoles from the marine blue-green alga Rivularia firma. Application of carbon-13 NMR spin-lattice relaxation data and carbon-13-proton coupling constants to structure elucidation. J. Am. Chem. Soc. 1982, 104, 3628–3635. [Google Scholar] [CrossRef]
- Kubota, N.K.; Iwamoto, H.; Fukuzawa, Y.; Uchio, Y. Five new sulfur-containing polybrominated bisindoles from the red alga Laurencia brongniartii. Heterocycles 2005, 65, 2675–2682. [Google Scholar]
- Liang, Z.; Zhao, J.; Zhang, Y. Palladium-catalyzed regioselective oxidative coupling of indoles and one-pot synthesis of acetoxylated biindolyls. J. Org. Chem. 2010, 75, 170–177. [Google Scholar] [CrossRef]
- Li, Y.-X.; Ji, K.-G.; Wang, H.-X.; Ali, S.; Liang, Y.-M. Iodine-induced regioseletive C–C and C–N bonds formation of N-protected indoles. J. Org. Chem. 2011, 76, 744–747. [Google Scholar] [CrossRef]
- Guo, T.; Han, S.-L.; Liu, Y.-C.; Liu, Y.; Liu, H.-M. Convenient synthesis of antiproliferative 2,3-dihydro-2,3′-bisindoles via dimerization of N–H indole derivatives. Tetrahedron Lett. 2016, 57, 1097–1099. [Google Scholar] [CrossRef]
- Huang, P.; Peng, X.; Hu, D.; Liao, H.; Tang, S.; Liu, L. Regioselective synthesis of 2,3′-biindoles mediated by an NBS-induced homo-coupling of indoles. Org. Biomol. Chem. 2017, 15, 9622–9629. [Google Scholar] [CrossRef]
- Yin, B.; Huang, P.; Lu, Y.; Liu, L. TEMPO-catalyzed oxidative homocoupling route to 3,2′-biindolin-2-ones via an indolin-3-one intermediate. RSC Adv. 2017, 7, 606–610. [Google Scholar] [CrossRef] [Green Version]
- Benkovics, T.; Guzei, I.A.; Yoon, T.P. Oxaziridine-mediated oxyamination of indoles: An approach to 3-aminoindoles and enantiomerically enriched 3-aminopyrroloindolines. Angew. Chem. Int. Ed. 2010, 49, 9153–9157. [Google Scholar] [CrossRef]
- Lee, D.J.; Yoo, E.J. Efficient synthesis of C–N-couped heterobiaryls by sequential N–H functionalization reactions. Org. Lett. 2015, 17, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-R.; Cheng, B.-Y.; Wang, Y.-N.; Zhang, M.-M.; Lu, L.-Q.; Xiao, W.-J. A copper-catalyzed decarboxylative amination/hydroamination sequence: Switchable synthesis of functionalized indoles. Angew. Chem. Int. Ed. 2016, 55, 12422–12426. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, K.; Yoshimura, Y.; Akehi, M.; Tsuchimoto, T. A heteroarylamine library: Indium-catalyzed nucleophilic aromatic substitution of alkoxyheteroarenes with amines. Adv. Synth. Catal. 2018, 360, 1159–1181. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Tian, J.-S.; Tan, P.-W.; Cao, Q.; Zhang, X.-X.; Cao, Z.-Y.; Zhou, F.; Wang, X.; Zhou, J. Ragiodivergent intramolecular nucleophilic addition of ketimines for the diverse synthesis of azacycles. Angew. Chem. Int. Ed. 2020, 59, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.-H.; Zheng, H.-X.; Yang, B.; Tie, L.; Fu, J.-L.; Qu, J.-P.; Kang, Y.-B. Copper-catalyzed oxidative benzylic C–H cyclization via iminyl radical from intermolecular anion-radical redox relay. Nat. Commun. 2019, 10, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
- Somei, M. A frontier in indole chemistry: 1-Hydroxyindoles, 1-hydroxytryptamines, and 1-hydrxytryptophans. Top. Heterocycl. Chem. 2006, 6, 77–111. [Google Scholar]
- Somei, M.; Yamada, F.; Hayashi, T.; Goto, A.; Saga, Y. Nucleophilic substitution reaction on the nitrogen of indole nucleus: Formation of 1-(indol-3-yl)indoles upon reaction of 1-hydroxyindoles with indole in formic acid. Heterocycles 2001, 55, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Peng, W.; Nakai, Y.; Yamada, K.; Somei, M. Nucleophilic substitution reaction on the nitrogen of indole nucleus: A novel synthesis of 1-aryltryptamines. Heterocycles 2002, 57, 421–424. [Google Scholar]
- Yamada, F.; Goto, A.; Peng, W.; Hayashi, T.; Saga, Y.; Somei, M. Nucleophilic substitution reaction at the 1-position of 1-hydroxytryptamine and -tryptophan derivatives. Heterocycles 2003, 61, 163–172. [Google Scholar]
- O’Neil, L.G.; Bower, J.F. Electrophilic aminating agents in total synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Kim, S.-T.; Jeong, J.; Baik, M.-H.; Buchwald, S.L. CuH-Catalyzed enantioselective alkylation of indole derivatives with ligand-controlled regiodivergence. J. Am. Chem. Soc. 2019, 141, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Suzuki, T.; Anada, M.; Matsunaga, S.; Yamada, K. 2-Hydroxyindoline-3-triethylammonium bromide: A reagent for formal C3-electrophilic reactions of indoles. Org. Lett. 2017, 19, 4275–4278. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Yamada, K. Dehydrative Mannich-type reaction for the synthesis of azepinobisindole alkaloid iheyamine A. Org. Lett. 2018, 20, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Shimizu, H.; Takada, S.; Tanaka, T.; Yoshikawa, M.; Yamada, K. Double “open and shut” transformation of g-carbolines triggered by ammonium salts: One-pot synthesis of multiheterocyclic compounds. Org. Lett. 2018, 20, 1589–1592. [Google Scholar] [CrossRef]
- Abe, T.; Satake, S.; Yamada, K. Biomimetic synthesis of iheyamine A from spirocyclic oxindoles. Heterocycles 2019, 99, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Aoyama, S.; Ohmura, M.; Taniguchi, M.; Yamada, K. Revisiting furodiindolines: One-pot synthesis of furodiindolines using indole 2,3-epoxide surrogates and their synthetic applications. Org. Lett. 2019, 21, 3367–3371. [Google Scholar] [CrossRef]
- Abe, T.; Yamada, K.; Nishi, T. Development and application of indole-2,3-epoxide surrogates. J. Synth. Org. Chem. 2020, 78, 597–607. [Google Scholar] [CrossRef]
- Abe, T.; Yamashiro, T.; Hirao, S. A metal-, oxidant-, and fluorous solvent-free synthesis of a-indolylketones enabled by an umpolung strategy. Chem. Commun. 2020, 56, 10183–10186. [Google Scholar] [CrossRef]
- Abe, T.; Noda, K.; Sawada, D. Synthesis of a-substituted indolylacetamide using acetonitriles as acetamide enolate equivalents through O-transfer reactions. Chem. Commun. 2021, 57, 7493–7496. [Google Scholar] [CrossRef]
- Yamashiro, T.; Abe, T.; Tanioka, M.; Kamino, S.; Sawada, D. cis-3-Azido-2-methoxyindolines as safe and stable precursors to overcome he instability of fleeting 3-azidoindoles. Chem. Commun. 2021, 57, 13381–13384. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Abe, T.; Sawada, D. Synthesis of 2-monosubstituted indolin-3-ones by cine-substitution of 3-azido-2-methoxyindolines. Org. Chem. Front. 2022, 9, 1897–1903. [Google Scholar] [CrossRef]
- Abe, T.; Yamashiro, T.; Shimizu, K.; Sawada, D. Indole editing enabled by HFIP-mediated ring-switch reactions of 3-amino-2-hydroxyindolines. Chem. Eur. J. 2022, 28, e202201113. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Nakajima, R.; Yamashiro, T.; Sawada, D. First total synthesis of reassigned echinosulfonic acid D. J. Nat. Prod. 2022, 85, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Hirao, S.; Yamashiro, T.; Kohira, K.; Mishima, N.; Abe, T. 2,3-Dimethoxyindolines: A latent electrophile for SNAr reactions triggered by indium catalysts. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef]
- Abe, T.; Hirao, S. Rapid access to indole-fused bicyclo[2.2.2]octanones by merging the umpolung strategy and molecular iodine as a green catalyst. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef]
- Sestelo, J.P.; Sarandeses, L.A.; Martínez, M.M.; Alonso-Marañón, L. Indium(III) as p-acid catalyst for the electrophilic activation of carbon-carbon unsaturated systems. Org. Biomol. Chem. 2018, 16, 5733–5747. [Google Scholar] [CrossRef]
- Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Organoindium reagents: The preparation and application in organic synthesis. Chem. Rev. 2013, 113, 271–401. [Google Scholar] [CrossRef]
- Zhao, K.; Shen, L.; Shen, Z.-L.; Loh, T.-P. Transition metal-catalyzed cross-coupling reactions using organoindium reagents. Chem. Rev. 2017, 46, 586–602. [Google Scholar] [CrossRef]
- Gohain, M.; Marais, C.; Bezuidenhoudt, C.B. An Al(OTf)3-catalyzed environmentally benign process for the propargylation of indoles. Tetrahedron Lett. 2012, 53, 4704–4707. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Alcohols in direct carbon-carbon and carbon-heteroatom bond-forming reactions: Recent advances. Arkivoc 2018, 2018, 288–329. [Google Scholar] [CrossRef] [Green Version]
- Bandini, M. Electrophilicity: The “dark-side” of indole chemistry. Org. Biomol. Chem. 2013, 11, 5206–5212. [Google Scholar] [CrossRef] [PubMed]
- Cerveri, A.; Bandini, M. Recent advances in the catalytic functionalization of “electrophilic” indoles. Chin. J. Chem. 2020, 38, 287–294. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kodama, A.; Nishida, T.; Shimizu, K.; Somei, M. Preparation of 1-hydroxyindole derivatives and a new route to 2-substituted indoles. Heterocycles 1991, 32, 221–227. [Google Scholar]
- Vo, Q.V.; Trenerry, C.; Rochfort, S.; Wadespm, J.; Leyton, C.; Hughes, A.B. Synthesis and anti-inflammatory activity of indole glucosinolates. Bioorg. Med. Chem. 2014, 22, 856–864. [Google Scholar] [CrossRef]
- Pezzella, A.; Panzella, L.; Natangelo, A.; Arzillo, M.; Napolitano, A.; d’Ischia, M. 5,6-Dihydroxyindole tetramers with “anomalous” interunit bonding patterns by oxidative coupling of 5,5′,6,6′-tetrahydroxy-2,7′-biindolyl: Emerging complexities on the way toward an improved model of eumelanin build up. J. Org. Chem. 2007, 72, 9225–9230. [Google Scholar] [CrossRef] [PubMed]
- Arzillo, M.; Pezzella, A.; Crescezi, O.; Napolitano, A.; Land, E.J.; Barone, V.; d’Ischia, M. Cyclic structural motifs in 5,6-dihydroxyindole polymerization uncovered: Biomimetic modular buildup of a unique five-membered macrocycle. Org. Lett. 2010, 12, 3250–3253. [Google Scholar] [CrossRef]
- Chen, C.-T.; Chuang, C.; Cao, J.; Ball, V.; Ruch, D.; Buehler, M.J. Excitonic effects from geometric order and disorder explain broadband optical absorption in eumelanin. Nat. Commun. 2014, 5, 3859. [Google Scholar] [CrossRef] [Green Version]
- Jamison, C.R.; Badillo, J.J.; Lipshultz, J.M.; Comito, R.J.; MacMillan, D.W.C. Catalyst-controlled oligomerization for the collective synthesis of polypyrroloindoline natural products. Nat. Chem. 2017, 9, 1165–1169. [Google Scholar] [CrossRef]
- Chang, K.-J.; Kang, B.-N.; Lee, M.-H.; Jeong, K.-S. Oligoindole-based foldamers with a helical conformation induced by chloride. J. Am. Chem. Soc. 2005, 127, 12214–12215. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, W.; Jiang, F.; Chang, Y.; Zhou, Q.; Li, D.; Ye, G.; Li, C.; Nie, G.; Xu, J.; et al. Three-dimensional porous carbon derived from polyindole hollow nanospheres for high-performance supercapacitor electrode. ACS Appl. Energy Mater. 2018, 1, 4572–4579. [Google Scholar] [CrossRef]
- Bürger, M.; Ehrhardt, N.; Barber, T.; Ball, L.T.; Namyslo, J.C.; Jones, P.G.; Werz, D.B. Phosphine-catalyzed aryne oligomerization: Direct access to a,w-bisfunctionalized oligo(ortho-arylenes). J. Am. Chem. Soc. 2021, 143, 16796–16803. [Google Scholar] [CrossRef] [PubMed]
- Boknevitz, K.; Darrigan, C.; Chrostowska, A.; Liu, S.-Y. Cation–p binding ability of BN indole. Chem. Commun. 2020, 56, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Wang, Y.; Wang, C.; Li, Y.; Xu, Y.; Yang, L. A recyclable hydroxyl functionalized polyindole hydrogel for sodium hydroxide extraction via the synergistic effect of cation-p interactions and hydrogen boning. Chem. Commun. 2018, 54, 9785–9788. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Run 1 | Catalyst | Solvent | Time (h) | Yield (%) of 3aa 2 |
| 1 | In(OTf)3 | MeCN | 1.5 | 72 |
| 2 | InF3·3H2O | MeCN | 1.5 | 14 |
| 3 | InBr3 | MeCN | 1.5 | 18 |
| 4 | InCl3·4H2O | MeCN | 1.5 | 8 |
| 5 | Ga(OTf)3 | MeCN | 1.5 | 79 |
| 6 | La(OTf)3 | MeCN | 1.5 | 31 |
| 7 | Bi(OTf)3 | MeCN | 1.5 | 60 |
| 8 | AgOTf | MeCN | 1.5 | 15 |
| 9 | Yb(OTf)3 | MeCN | 1.5 | 0 |
| 10 | Cu(OTf)2 | MeCN | 1.5 | 72 |
| 11 | Zn(OTf)2 | MeCN | 1.5 | 7 |
| 12 | Al(OTf)3 | MeCN | 1.5 | 83 (87) 3 |
| 13 | AlCl3 | MeCN | 1.5 | 23 |
| 14 | Al(O-iPr)3 | MeCN | 1.5 | 9 |
| 15 | Al(OTf)3 | PhCl | 1.5 | 69 |
| 16 | Al(OTf)3 | 1,4-dioxane | 1.5 | 43 |
| 17 | Al(OTf)3 | CHCl3 | 1.5 | 71 |
| 18 | TfOH | MeCN | 1.5 | 54 |
| 19 | --- | MeCN | 24 | nr |
| 20 | Al(OTf)3 | --- | 24 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokushige, K.; Yamashiro, T.; Hirao, S.; Abe, T. Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry 2023, 5, 452-462. https://doi.org/10.3390/chemistry5010033
Tokushige K, Yamashiro T, Hirao S, Abe T. Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry. 2023; 5(1):452-462. https://doi.org/10.3390/chemistry5010033
Chicago/Turabian StyleTokushige, Keisuke, Toshiki Yamashiro, Seiya Hirao, and Takumi Abe. 2023. "Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles" Chemistry 5, no. 1: 452-462. https://doi.org/10.3390/chemistry5010033
APA StyleTokushige, K., Yamashiro, T., Hirao, S., & Abe, T. (2023). Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry, 5(1), 452-462. https://doi.org/10.3390/chemistry5010033