


Crotonaldehyde Adsorption on Cu-Pt Surface Alloys: A Quantum Mechanics Study


Abstract
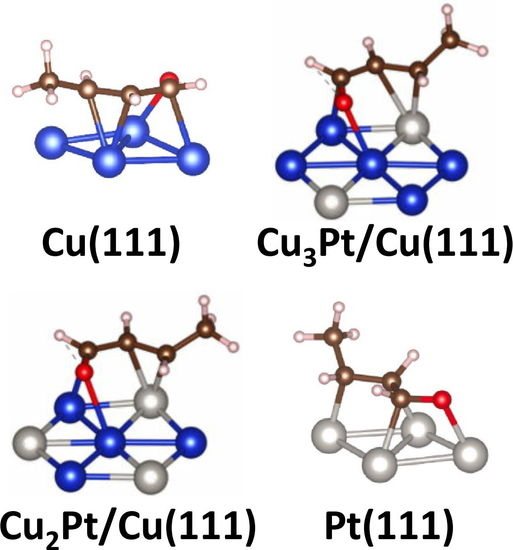
:
1. Introduction
2. Methods
3. Results
3.1. Energetics and Structural Properties
3.2. Vibrational Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bond, G.C. Metal-Catalysed Reactions of Hydrocarbons; Springer: New York, NY, USA, 2005. [Google Scholar]
- Sanfilippo, D.; Rylander, P.N. Hydrogenation and Dehydrogenation. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; Volume 18, pp. 451–471. [Google Scholar]
- Ma, Z.; Zaera, F. Heterogeneous Catalysis by Metals. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; p. eibc0079. [Google Scholar]
- Sinfelt, J.H. Bimetallic Catalysts: Discoveries, Concepts and Applications; John Wiley and Sons: New York, NY, USA, 1983; p. 164. [Google Scholar]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef]
- Yu, W.; Porosoff, M.D.; Chen, J.G. Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts. Chem. Rev. 2012, 112, 5780–5817. [Google Scholar] [CrossRef]
- Meemken, F.; Baiker, A. Recent Progress in Heterogeneous Asymmetric Hydrogenation of C═O and C═C Bonds on Supported Noble Metal Catalysts. Chem. Rev. 2017, 117, 11522–11569. [Google Scholar] [CrossRef]
- Zaera, F. The Surface Chemistry of Metal-Based Hydrogenation Catalysis. ACS Catal. 2017, 7, 4947–4967. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Almora-Barrios, N.; López, N.; Pérez-Ramírez, J. Advances in the Design of Nanostructured Catalysts for Selective Hydrogenation. ChemCatChem 2016, 8, 21–33. [Google Scholar] [CrossRef]
- Burch, R.; Garla, L.C. Platinum-tin reforming catalysts: II. Activity and selectivity in hydrocarbon reactions. J. Catal. 1981, 71, 360–372. [Google Scholar] [CrossRef]
- Joyner, R.W.; Shpiro, E.S. Alloying in platinum-based catalysts for gasoline reforming: A general structural proposal. Catal. Lett. 1991, 9, 239–243. [Google Scholar] [CrossRef]
- Delbecq, F.; Sautet, P. Influence of Sn additives on the selectivity of hydrogenation of α-β-unsaturated aldehydes with Pt catalysts: A density functional study of molecular adsorption. J. Catal. 2003, 220, 115–126. [Google Scholar] [CrossRef]
- Yang, X.; Koel, B.E. Adsorption and Reaction of Unsaturated Hydrocarbons on Sn/Pt Alloys. In Encyclopedia of Interfacial Chemistry; Wandelt, K., Ed.; Elsevier: Oxford, UK, 2018; pp. 1–10. [Google Scholar]
- Stassi, J.; Méndez, J.; Vilella, I.; de Miguel, S.; Zgolicz, P. Synthesis of PtSn nanoparticles on carbon materials by different preparation methods for selective catalytic hydrogenation of citral. Chem. Eng. Commun. 2020, 207, 1074–1091. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, B.; Guerrero-Sánchez, J.; Lee, I.; Zhou, X.; Takeuchi, N.; Zaera, F. Controlling Selectivity in Unsaturated Aldehyde Hydrogenation Using Single-Site Alloy Catalysts. ACS Catal. 2019, 9, 9150–9157. [Google Scholar] [CrossRef]
- Chen, B.; Zaera, F. Hydrogenation of Cinnamaldehyde on Cu(110) Single-Crystal Surfaces. J. Phys. Chem. C 2021, 125, 14709–14717. [Google Scholar] [CrossRef]
- Nayakasinghe, M.T.; Ponce Perez, R.; Chen, B.; Takeuchi, N.; Zaera, F. Adsorption, thermal conversion, and catalytic hydrogenation of acrolein on Cu surfaces. J. Catal. 2022, 414, 257–266. [Google Scholar] [CrossRef]
- Johansson, M.; Lytken, O.; Chorkendorff, I. The sticking probability for H2 on some transition metals at a hydrogen pressure of 1 bar. J. Chem. Phys. 2008, 128, 034706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Falcón, L.; Viñes, F.; Notario-Estévez, A.; Illas, F. On the hydrogen adsorption and dissociation on Cu surfaces and nanorows. Surf. Sci. 2016, 646, 221–229. [Google Scholar] [CrossRef]
- Han, J.; Lu, J.; Wang, M.; Wang, Y.; Wang, F. Single Atom Alloy Preparation and Applications in Heterogeneous Catalysis. Chin. J. Chem. 2019, 37, 977–988. [Google Scholar] [CrossRef]
- Hannagan, R.T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-Atom Alloy Catalysis. Chem. Rev. 2020, 120, 12044–12088. [Google Scholar] [CrossRef]
- Zaera, F. Molecular approaches to heterogeneous catalysis. Coord. Chem. Rev. 2021, 448, 214179. [Google Scholar] [CrossRef]
- Zaera, F. Designing Sites in Heterogeneous Catalysis: Are We Reaching Selectivities Competitive with Those of Homogeneous Catalysts? Chem. Rev. 2022, 122, 8594–8757. [Google Scholar] [CrossRef]
- Luneau, M.; Lim, J.S.; Patel, D.A.; Sykes, E.C.H.; Friend, C.M.; Sautet, P. Guidelines to Achieving High Selectivity for the Hydrogenation of α,β-Unsaturated Aldehydes with Bimetallic and Dilute Alloy Catalysts: A Review. Chem. Rev. 2020, 120, 12834–12872. [Google Scholar] [CrossRef]
- Sykes, E.C.H.; Christopher, P. Recent advances in single-atom catalysts and single-atom alloys: Opportunities for exploring the uncharted phase space in-between. Curr. Opin. Chem. Eng. 2020, 29, 67–73. [Google Scholar] [CrossRef]
- Thirumalai, H.; Kitchin, J.R. Investigating the Reactivity of Single Atom Alloys Using Density Functional Theory. Top. Catal. 2018, 61, 462–474. [Google Scholar] [CrossRef]
- Schumann, J.; Bao, Y.; Hannagan, R.T.; Sykes, E.C.H.; Stamatakis, M.; Michaelides, A. Periodic Trends in Adsorption Energies around Single-Atom Alloy Active Sites. J. Phys. Chem. Lett. 2021, 12, 10060–10067. [Google Scholar] [CrossRef] [PubMed]
- Zaera, F. In-Situ and Operando Spectroscopies for the Characterization of Catalysts and of Mechanisms of Catalytic Reactions. J. Catal. 2021, 404, 900–910. [Google Scholar] [CrossRef]
- Zafeiratos, S.; Piccinin, S.; Teschner, D. Alloys in catalysis: Phase separation and surface segregation phenomena in response to the reactive environment. Catal. Sci. Technol. 2012, 2, 1787–1801. [Google Scholar] [CrossRef] [Green Version]
- Gumuslu, G.; Kondratyuk, P.; Boes, J.R.; Morreale, B.; Miller, J.B.; Kitchin, J.R.; Gellman, A.J. Correlation of Electronic Structure with Catalytic Activity: H2–D2 Exchange across CuxPd1–x Composition Space. ACS Catal. 2015, 5, 3137–3147. [Google Scholar] [CrossRef]
- Simonovis, J.P.; Hunt, A.; Palomino, R.M.; Senanayake, S.D.; Waluyo, I. Enhanced Stability of Pt-Cu Single-Atom Alloy Catalysts: In Situ Characterization of the Pt/Cu(111) Surface in an Ambient Pressure of CO. J. Phys. Chem. C 2018, 122, 4488–4495. [Google Scholar] [CrossRef]
- Yang, K.; Yang, B. Identification of the Active and Selective Sites over a Single Pt Atom-Alloyed Cu Catalyst for the Hydrogenation of 1,3-Butadiene: A Combined DFT and Microkinetic Modeling Study. J. Phys. Chem. C 2018, 122, 10883–10891. [Google Scholar] [CrossRef]
- Han, T.; Li, Y.; Cao, Y.; Lee, I.; Zhou, X.; Frenkel, A.I.; Zaera, F. In situ identification of surface sites in Cu–Pt bimetallic catalysts: Gas-induced metal segregation. J. Chem. Phys. 2022, 157, 234706. [Google Scholar] [CrossRef]
- Finzel, J.; Christopher, P. Dynamic Pt Coordination in Dilute AgPt Alloy Nanoparticle Catalysts Under Reactive Environments. Top. Catal. 2022, 65, 1587–1603. [Google Scholar] [CrossRef]
- Rodriguez, J.A. Physical and Chemical Properties of Bimetallic Surfaces. Surf. Sci. Rep. 1996, 24, 223–287. [Google Scholar] [CrossRef]
- Wang, Y.; Balbuena, P.B. Design of Oxygen Reduction Bimetallic Catalysts: Ab-Initio-Derived Thermodynamic Guidelines. J. Phys. Chem. B 2005, 109, 18902–18906. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Greiner, M.T.; Jones, T.E.; Beeg, S.; Zwiener, L.; Scherzer, M.; Girgsdies, F.; Piccinin, S.; Armbrüster, M.; Knop-Gericke, A.; Schlögl, R. Free-atom-like d states in single-atom alloy catalysts. Nat. Chem. 2018, 10, 1008–1015. [Google Scholar] [CrossRef]
- Duchesne, P.N.; Li, Z.Y.; Deming, C.P.; Fung, V.; Zhao, X.; Yuan, J.; Regier, T.; Aldalbahi, A.; Almarhoon, Z.; Chen, S.; et al. Golden single-atomic-site platinum electrocatalysts. Nat. Mater. 2018, 17, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Hopper, N.; Thuening, T.; Manzi, S.; Weinert, M.; Tysoe, W.T. Binding of Oxygen on Single-Atom Sites on Au/Pd(100) Alloys with High Gold Coverages. J. Phys. Chem. C 2021, 125, 9715–9729. [Google Scholar] [CrossRef]
- Xin, H.; Vojvodic, A.; Voss, J.; Nørskov, J.K.; Abild-Pedersen, F. Effects of d-band shape on the surface reactivity of transition-metal alloys. Phys. Rev. B 2014, 89, 115114. [Google Scholar] [CrossRef]
- Han, T.; Lee, I.; Cao, Y.; Zhou, X.; Zaera, F. Thermodynamics of Carbon Monoxide Adsorption on Cu/SBA-15 Catalysts: Under Vacuum versus under Atmospheric Pressures. J. Phys. Chem. C 2022, 126, 3078–3086. [Google Scholar] [CrossRef]
- Ponec, V. Alloy catalysts: The concepts. Appl. Catal. A 2001, 222, 31–45. [Google Scholar] [CrossRef]
- Liu, P.; Nørskov, J.K. Ligand and ensemble effects in adsorption on alloy surfaces. Phys. Chem. Chem. Phys. 2001, 3, 3814–3818. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. Pd-Au bimetallic catalysts: Understanding alloy effects from planar models and (supported) nanoparticles. Chem. Soc. Rev. 2012, 41, 8009–8020. [Google Scholar] [CrossRef] [PubMed]
- Trimm, D.L. Coke Formation and Minimisation During Steam Reforming Reactions. Catal. Today 1997, 37, 233–238. [Google Scholar] [CrossRef]
- Marcella, N.; Lim, J.S.; Płonka, A.M.; Yan, G.; Owen, C.J.; van der Hoeven, J.E.S.; Foucher, A.C.; Ngan, H.T.; Torrisi, S.B.; Marinkovic, N.S.; et al. Decoding reactive structures in dilute alloy catalysts. Nat. Commun. 2022, 13, 832. [Google Scholar] [CrossRef]
- Mancera, L.A.; Diemant, T.; Groß, A.; Behm, R.J. Molecular and Dissociative Hydrogen Adsorption on Bimetallic PdAg/Pd(111) Surface Alloys: A Combined Experimental and Theoretical Study. J. Phys. Chem. C 2022, 126, 3060–3077. [Google Scholar] [CrossRef]
- Lucci, F.R.; Liu, J.; Marcinkowski, M.D.; Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Selective hydrogenation of 1,3-butadiene on platinum–copper alloys at the single-atom limit. Nat. Commun. 2015, 6, 8550. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.P.; Chen, J.G. Correlating hydrogenation activity with binding energies of hydrogen and cyclohexene on M/Pt(111) (M = Fe, Co, Ni, Cu) bimetallic surfaces. J. Catal. 2008, 257, 297–306. [Google Scholar] [CrossRef]
- Lv, C.-Q.; Liu, J.-H.; Guo, Y.; Wang, G.-C. Selective hydrogenation of 1,3-butadiene over single Pt1/Cu(111) model catalysts: A DFT study. Appl. Surf. Sci. 2019, 466, 946–955. [Google Scholar] [CrossRef]
- Liu, D.; Chen, H.Y.; Zhang, J.Y.; Huang, J.Y.; Li, Y.M.; Peng, Q.M. Theoretical investigation of selective hydrogenation of 1,3-butadiene on Pt doping Cu nanoparticles. Appl. Surf. Sci. 2018, 456, 59–68. [Google Scholar] [CrossRef]
- Cao, Y.; Guerrero-Sańchez, J.; Lee, I.; Zhou, X.; Takeuchi, N.; Zaera, F. Kinetic Study of the Hydrogenation of Unsaturated Aldehydes Promoted by CuPtx/SBA-15 Single-Atom Alloy (SAA) Catalysts. ACS Catal. 2020, 10, 3431–3443. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Vanderbilt, D.; Hamann, D.R. Systematic treatment of displacements, strains, and electric fields in density-functional perturbation theory. Phys. Rev. B 2005, 72, 035105. [Google Scholar] [CrossRef] [Green Version]
- Karhánek, D. Dakarhanek/Vasp-Infrared-Intensities: Vasp-Infrared-Intensities; (Version v1.0); Zenodo, 2020; Available online: https://zenodo.org/record/3930989#.ZAF24R9ByUk (accessed on 26 January 2023).
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Nayakasinghe, M.T.; Guerrero-Sánchez, J.; Takeuchi, N.; Zaera, F. Adsorption of crotonaldehyde on metal surfaces: Cu vs Pt. J. Chem. Phys. 2021, 154, 104701. [Google Scholar] [CrossRef]
- Haubrich, J.; Becker, C.; Wandelt, K. Adsorption and interaction energy of π ethene on Pt(111) and Pt alloys: A detailed analysis of vibrational, energetic and electronic properties. Surf. Sci. 2009, 603, 1476–1485. [Google Scholar] [CrossRef]
- Lindenmaier, R.; Williams, S.D.; Sams, R.L.; Johnson, T.J. Quantitative Infrared Absorption Spectra and Vibrational Assignments of Crotonaldehyde and Methyl Vinyl Ketone Using Gas-Phase Mid-Infrared, Far-Infrared, and Liquid Raman Spectra: S-cis vs s-trans Composition Confirmed via Temperature Studies and ab Initio Methods. J. Phys. Chem. A 2017, 121, 1195–1212. [Google Scholar] [CrossRef]
- de Jesús, J.C.; Zaera, F. Adsorption and Thermal Chemistry of Acrolein and Crotonaldehyde on Pt(111) Surfaces. Surf. Sci. 1999, 430, 99–115. [Google Scholar] [CrossRef]
- Murillo, L.E.; Menning, C.A.; Chen, J.G. Trend in the CC and CO bond hydrogenation of acrolein on Pt–M (M = Ni, Co, Cu) bimetallic surfaces. J. Catal. 2009, 268, 335–342. [Google Scholar] [CrossRef]
- de Jesús, J.C.; Zaera, F. Double-Bond Activation in Unsaturated Aldehydes: Conversion of Acrolein to Propene and Ketene on Pt(111) Surfaces. J. Mol. Catal. A Chem. 1999, 138, 237–240. [Google Scholar] [CrossRef]
- Islam, A.; Molina, D.L.; Trenary, M. Adsorption of acrolein and its hydrogenation products on Cu(111). Phys. Chem. Chem. Phys. 2022, 24, 24383–24393. [Google Scholar] [CrossRef] [PubMed]
- Greenler, R.G. Infrared Study of Adsorbed Molecules on Metal Surfaces by Reflection Techniques. J. Chem. Phys. 1966, 44, 310–315. [Google Scholar] [CrossRef]
- Zaera, F. New advances in the use of infrared absorption spectroscopy for the characterization of heterogeneous catalytic reactions. Chem. Soc. Rev. 2014, 43, 7624–7663. [Google Scholar] [CrossRef]
- Chen, B.; Ponce, R.; Guerrero-Sánchez, J.; Takeuchi, N.; Zaera, F. Cinnamaldehyde adsorption and thermal decomposition on copper surfaces. J. Vac. Sci. Technol. A 2021, 39, 053205. [Google Scholar] [CrossRef]
- Lucci, F.R.; Darby, M.T.; Mattera, M.F.G.; Ivimey, C.J.; Therrien, A.J.; Michaelides, A.; Stamatakis, M.; Sykes, E.C.H. Controlling Hydrogen Activation, Spillover, and Desorption with Pd–Au Single-Atom Alloys. J. Phys. Chem. Lett. 2016, 7, 480–485. [Google Scholar] [CrossRef]
- Fan, T.; Sun, M.; Ji, Y. First-principles study on the selective hydrogenation of the C=O and C=C bonds of acrolein on Pt–M–Pt (M = Pt, Cu, Ni, Co) surfaces. Phys. Chem. Chem. Phys. 2020, 22, 14645–14650. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
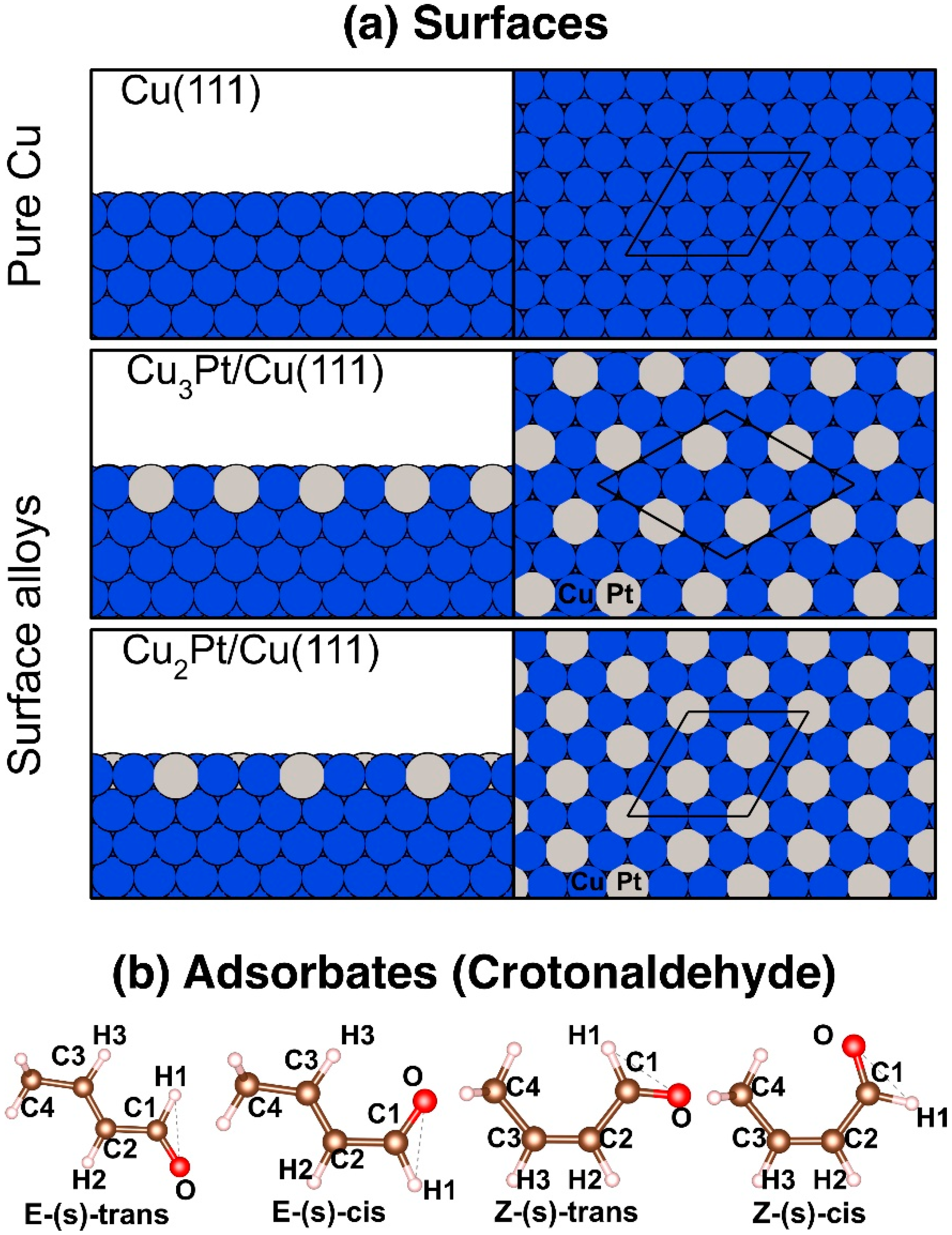{kind=link}
{kind=link}
{kind=link}
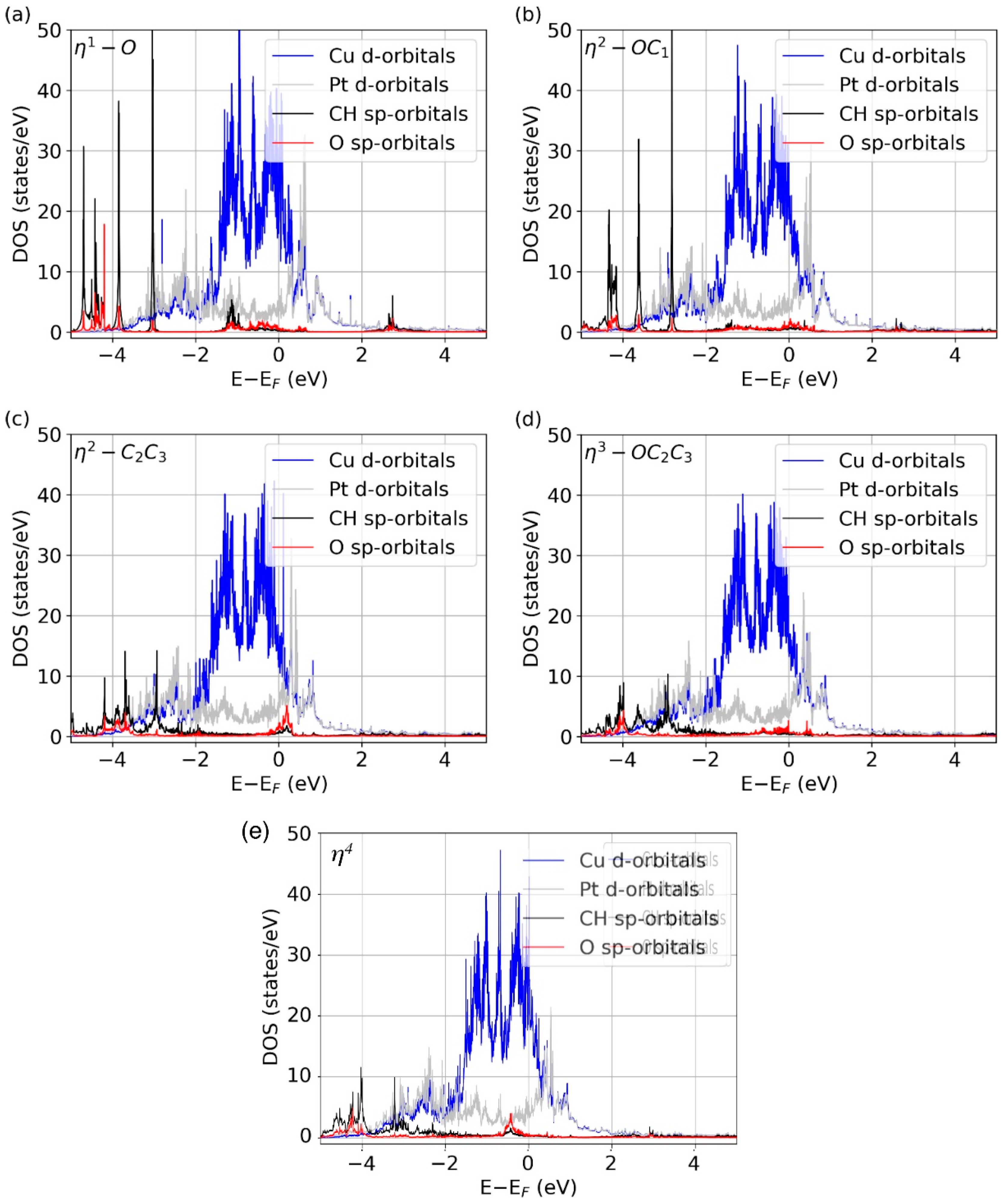{kind=link}
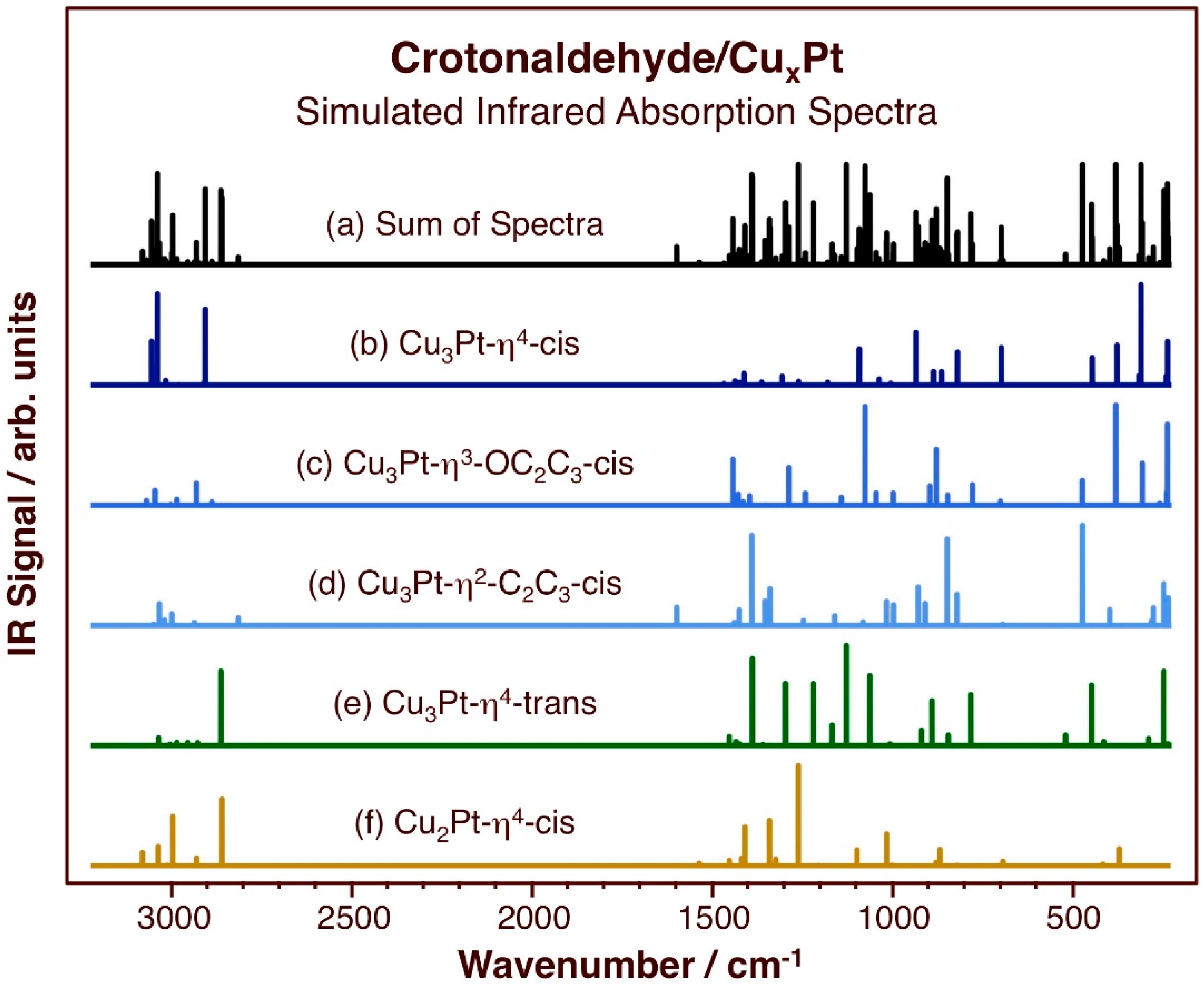{kind=link}
| Geometry | Gas Phase | Cu2Pt/Cu(111) | Cu3Pt/Cu(111) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E-(s)-cis | η1-O | η2-OC1 | η2-C2C3 | η3-OC2C3 | η4-OC1C2C3 | η1-O | η2-OC1 | η2-C2C3 | η3-OC2C3 | η4-OC1C2C3 | |
| Supercell | - | 3 × 3 | 3 × 3 | 3 × 3 | 3 × 3 | 3 × 3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 |
| Eab (kJ/mol) | - | −49.7 | −69.57 | −85.0 | −85.7 | −94.8 | −53.3 | −78.6 | −92.850 | −99.8 | −110.5 |
| d(C1=O) | 1.227 | 1.247 | 1.287 | 1.240 | 1.270 | 1.252 | 1.250 | 1.307 | 1.236 | 1.278 | 1.273 |
| d(C1–C2) | 1.474 | 1.455 | 1.461 | 1.461 | 1.434 | 1.462 | 1.453 | 1.465 | 1.470 | 1.425 | 1.443 |
| d(C2=C3) | 1.346 | 1.348 | 1.355 | 1.428 | 1.445 | 1.418 | 1.352 | 1.359 | 1.450 | 1.457 | 1.431 |
| d(C3–C4) | 1.486 | 1.482 | 1.485 | 1.512 | 1.512 | 1.502 | 1.483 | 1.485 | 1.514 | 1.514 | 1.509 |
| d(O–Cu/Pt) | – | 2.304 | 2.101 | – | 2.285 | 2.538 | 2.236 | 2.032 | – | 2.149 | 2.258 |
| d(C1–Cu/Pt) | – | – | 2.430 | – | – | 2.645 | – | 2.299 | 2.655 | – | 2.435 |
| d(C2–Cu/Pt) | – | – | – | 2.416 | 2.277 | 2.277 | – | – | 2.226 | 2.212 | 2.456 |
| d(C3–Cu/Pt) | – | – | – | 2.229 | 2.201 | 2.242 | – | – | 2.180 | 2.188 | 2.182 |
| Geometry | Gas Phase | Cu2Pt/Cu(111) | Cu3Pt/Cu(111) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E-(s)-trans | η1-O | η2-OC1 | η2-C2C3 | η3-OC2C3 | η4-OC1C2C3 | η1-O | η2-OC1 | η2-C2C3 | η3-OC2C3 | η4-OC1C2C3 | |
| Supercell | - | 3 × 3 | 3 × 3 | 3 × 3 | 3 × 3 | 3 × 3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 |
| Eab (kJ/mol) | - | −68.7 | −75.5 | −84.2 | −43.0 | −72.8 | −73.1 | −81.2 | −88.3 | −62.2 | −97.7 |
| d(C1=O) | 1.226 | 1.247 | 1.262 | 1.240 | 1.270 | 1.252 | 1.242 | 1.296 | 1.237 | 1.243 | 1.275 |
| d(C1–C2) | 1.463 | 1.455 | 1.457 | 1.461 | 1.434 | 1.462 | 1.444 | 1.464 | 1.463 | 1.474 | 1.434 |
| d(C2=C3) | 1.346 | 1.348 | 1.353 | 1.428 | 1.445 | 1.418 | 1.349 | 1.359 | 1.446 | 1.466 | 1.440 |
| d(C3–C4) | 1.488 | 1.482 | 1.486 | 1.512 | 1.512 | 1.502 | 1.484 | 1.483 | 1.516 | 1.512 | 1.511 |
| d(O–Cu/Pt) | – | 2.427 | 2.280 | – | 2.323 | 3.087 | 2.198 | 2.069 | – | 2.234 | 2.148 |
| d(C1–Cu/Pt) | – | – | 2.604 | – | – | 2.898 | – | 2.330 | – | – | 2.550 |
| d(C2–Cu/Pt) | – | – | – | 2.340 | 2.145 | 2.815 | – | – | 2.247 | 2.142 | 2.428 |
| d(C3–Cu/Pt) | – | – | – | 2.233 | 2.217 | 2.978 | – | – | 2.191 | 2.161 | 2.178 |
| Geometry | Gas phase | Cu3Pt/Cu(111) | Cu2Pt/Cu(111) | ||||
|---|---|---|---|---|---|---|---|
| E-(s)-trans | E-(s)-cis | η2-C2C3-cis | η3-OC2C3-cis | η4-OC1C2C3-cis | η4-OC1C2C3-trans | η4-OC1C2C3-cis | |
| Supercell | - | - | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 2√3 × 2√3 | 3 × 3 |
| ν(C–H) range | 3110-2753 | 3095–2828 | 3037–2876 | 3045–2880 | 3043–2888 | 3022–2859 | 3089–2863 |
| ν(C1=O) | 1701 | 1708 | |||||
| ν(C2=C3) | 1648 | 1622 | 1594 | 1537 | |||
| δa(CH3) | 1457 | 1473 | 1456 | 1462 | |||
| δ(CHx) | 1431, 1427 | 1423, 1429 | 1432, 1441 | 1423, 1435 | 1428, 1443 | 1427, 1440 | 1422, 1435 |
| δip(CH)ald | 1393 | 1400 | 1413 | 1396 | |||
| δs(CH3) | 1355, 1365 | 1351, 1376 | 1348, 1356 | 1356 | 1363 | 1358 | 1334, 1358 |
| δip(CH)vinyl | 1234, 1293 | 1271, 1288 | 1265 | 1245, 1283 | 1266, 1308 | 1223, 1293 | 1201, 1268 |
| ν(C1–C2) | 1136 | 1119 | 1148 | 1145 | 1182 | 1128, 1166 | 1110 |
| ν(C3–C4) | 1073 | 1080 | 1049, 1080 | 1092 | 1066 | ||
| 1030 | 1005, 1036 | 1015 | 999 | 1005 | 1003 | 998, 1025 | |
| ρ(CH3), τC2H2) | 973, 996 | 972, 989 | 985 | 947 | 963 | ||
| δoop(CH)ald | 926, 934 | 884, 904 | 823, 894 | 899, 920 | 913 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruvalcaba, R.; Guerrero-Sanchez, J.; Takeuchi, N.; Zaera, F. Crotonaldehyde Adsorption on Cu-Pt Surface Alloys: A Quantum Mechanics Study. Chemistry 2023, 5, 463-478. https://doi.org/10.3390/chemistry5010034
Ruvalcaba R, Guerrero-Sanchez J, Takeuchi N, Zaera F. Crotonaldehyde Adsorption on Cu-Pt Surface Alloys: A Quantum Mechanics Study. Chemistry. 2023; 5(1):463-478. https://doi.org/10.3390/chemistry5010034
Chicago/Turabian StyleRuvalcaba, Ricardo, Jonathan Guerrero-Sanchez, Noboru Takeuchi, and Francisco Zaera. 2023. "Crotonaldehyde Adsorption on Cu-Pt Surface Alloys: A Quantum Mechanics Study" Chemistry 5, no. 1: 463-478. https://doi.org/10.3390/chemistry5010034
APA StyleRuvalcaba, R., Guerrero-Sanchez, J., Takeuchi, N., & Zaera, F. (2023). Crotonaldehyde Adsorption on Cu-Pt Surface Alloys: A Quantum Mechanics Study. Chemistry, 5(1), 463-478. https://doi.org/10.3390/chemistry5010034