2. Materials and Methods
2.1. General Experimental
This section is part of the Ph.D. thesis submitted by G.B. [
22].
1H,
13C, NMR spectra were recorded using a Varian AS 300 and Bruker 400 and 600 spectrometer.
1H- and
13C- NMR for compounds
4–
18 can be found in
Supplementary Materials. Chemical shifts (δ) are reported in ppm relative to residual solvent signals for
1H and
13C NMR (1H NMR: 7.26 ppm for CDCl
3;
13C NMR: 77.0 ppm for CDCl
3.
13C NMR spectra were acquired using
1H broadband decoupled mode. DMSO-d6 was used (referenced to 2.52 and 3.35 ppm for 1H and 40.0 for
13C). Coupling constants (
J) are in Hz. Multiplicities are reported as follows: s—singlet; d—doublet; dd—doublets of doublets; t—triplet; q,—quartet; m—multiplet; c—complex; and br—broad.
1H-NMR spectral assignments are supported by
1H-
1H COSY and
13C-
1H-COSY where necessary. Carbon spectra are supported by DEPT analysis where necessary. Infrared spectra (IR) were obtained in CCl
4 using a Bruker Tensor 27 FT-IR instrument. Absorption maximum (ν
max) was reported in wave numbers (cm
−1) and only selected peaks are reported. High resolution mass spectra were obtained using a Waters Micro mass LCT and low-resolution mass spectra were recorded using Waters Micro mass Quattro LCMS spectrometers at 70 eV. Optical rotations were measured on a Perkin-Elmer 241 polarimeter. Tetrahydrofuran was freshly distilled over sodium benzophenone prior to use according to standard procedure. All other reagents and solvents were used as purchased from Aldrich. Reactions were checked for completion using TLC (EM Science, silica gel 60 F254), which were visualized by quenching of u.v. fluorescence (λ
max = 254 nm) or by staining with either 10%
w/
v ammonium molybdate in 2M sulfuric acid or basic potassium permanganate solution (followed by heat) as appropriate. Flash chromatography was performed using silica gel 60 (0.040–0.063 mm, 230–400 mesh). Retention factors (R
f) are reported to ±0.05.
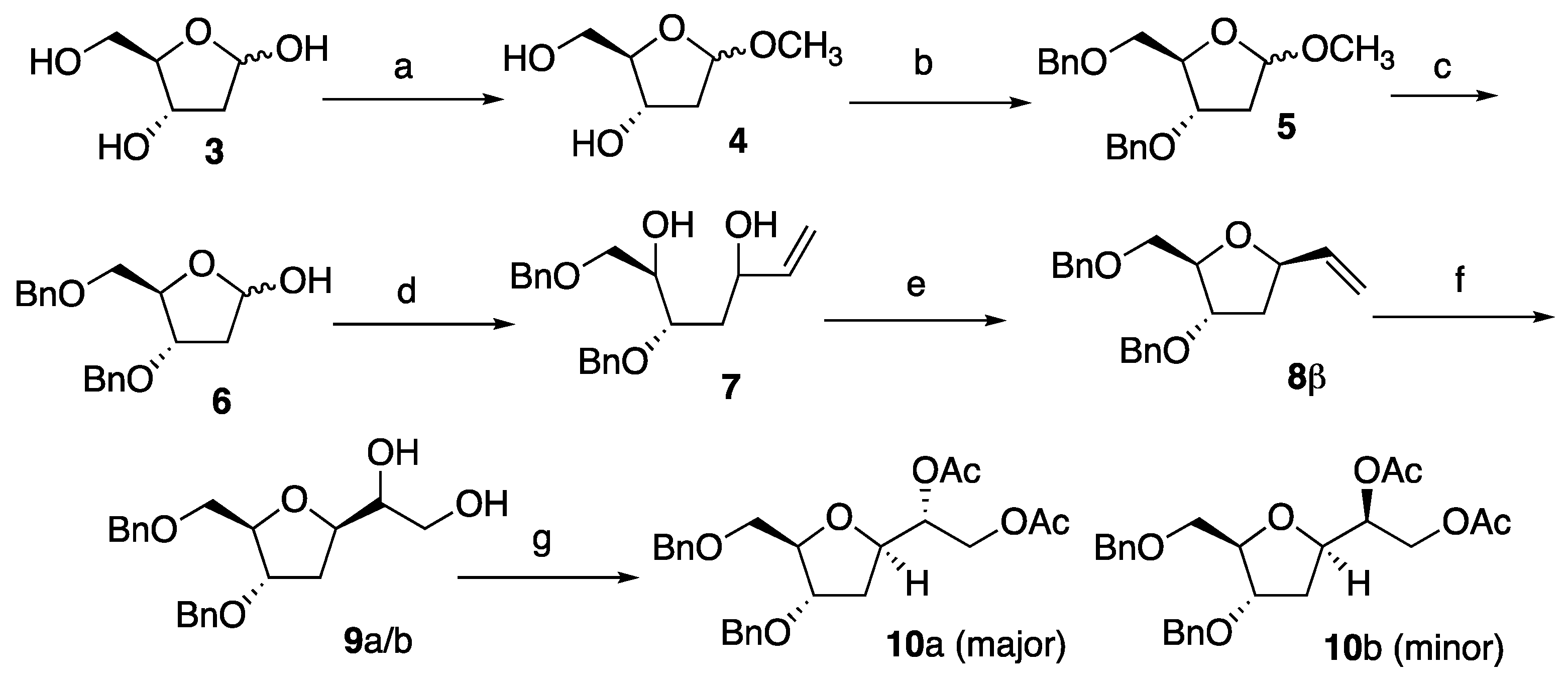
2.2. Synthesis of (2R,3S)-2-(Hydroxymethyl)-5-methoxytetrahydrofuran-3-ol 4
Acetyl chloride (690 μL, 6.5 mol%) was added To a stirred solution of 2-deoxy-D-ribose (20.0 g, 149.0 mmol) in methanol (240 mL). The reaction mixture was stirred at room temperature for 1 h; then, sodium bicarbonate (7.70 g) was added and the reaction stirred for a further 10 min. The solid formed was filtered through celite, and the filtrate was evaporated in vacuo to afford 4 as a mixture of two diastereoisomers as an orange oil (22.0 g, >99% yield). This product did not require any further purification. (α + β anomers): Rf = 0.53 (chloroform/methanol 8:2). δH (400 MHz, CDCl3): 5.19–4.95 (m, 2H), 4.39–4.32 (m, 1H), 4.11–4.08 (m, 1H), 3.99 (q, J = 4.4, 1H) 3.92 (q, J = 4.4, 1H), 3.70–3.55 (m, 4H), 3.32 (s, 3H, -CH3), 3.30 (s, 3H, -CH3), 2.19–2.10 (m, 2H), 2.06–2.00 (m, 1H), 1.92–1.88 (m, 1H). δC (100.6 MHz, CDCl3): 105.60, 105.56, 87.7, 87.5, 72.9, 72.3, 63.6, 63.2, 55.5, 54.9, 42.7, 41.6. HRMS (ESI): calculated for [M + Na]+, C6H12O4Na: 171.0633; found: 171.0638.
2.3. Synthesis of (2R,3S)-3-(Benzyloxy)-2-((benzyloxy)methyl)-5-methoxytetrahydrofuran 5
The reaction was split in two round-bottom flasks. To a stirred solution of 4 (22.7 g, 153.4 mmol) in THF (160 mL), powdered KOH (77.0 g, 1380.0 mmol, 9.0 eq.), and benzyl chloride (247.0 mL, 2148.0 mmol, 14.0 eq.) were added sequentially, and the reaction mixture was heated to reflux conditions for 24 h. The reaction mixture was allowed to cool to room temperature; then, the solution was filtered, and the solvent removed in vacuo. The residue was purified using flash chromatography on silica gel eluting the first time with petroleum ether to eliminate excess benzyl chloride, the second time with petroleum ether/ethyl acetate 8:2 to afford the title compound 5 as a yellow oil (42.6 g, 85% yield); (α + β anomers): Rf = 0.39 and 0.57 (petroleum ether/ethyl acetate 8:2). δH (400 MHz, CDCl3): 7.45–7.20 (m, 20H), 5.13–5.07 (m, 2H), 4.63–4.47 (m, 8H), 4.29–4.21 (m, 2H), 4.16–4.12 (m, 1H), 4.00–3.92 (m, 1H), 3.57–3.43 (m, 4H), 3.41 (s, 3H, -CH3), 3.31 (s, 3H, -CH3), 2.26–2.19 (m, 2H), 2.19–2.15 (m, 1H), 2.05–2.00 (m, 1H). δC (100.6 MHz, CDCl3):138.3, 138.23, 138.20, 138.1, 128.5, 128.4, 127.9, 127.8, 127.7, 127.6, 105.5, 105.3, 82.9, 82.2, 80.0, 78.6, 73.5, 73.4, 72.1, 71.7, 71.6, 70.2, 55.3, 55.0, 39.5, 39.0. HRMS (ESI): calculated for [M + Na]+, C20H24NaO4: 351.1572; found: 351.1568.
2.4. Synthesis of (4S,5R)-4-(Benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-ol 6
The reaction was split in two round-bottom flasks. A stirred solution of 5 (25.0 g, 76.0 mmol) in AcOH/H2O 80:20 (740 mL) was heated to 49 °C (external temperature) for 24 h. A solution of AcOH/H2O (80:20, 500 mL) was then added, and the reaction mixture was allowed to stir at the same temperature for another 24 h. The reaction was cooled to room temperature and the solvent was removed in vacuo. Heptane was added to the resulting crude mixture and then removed under reduced pressure to eliminate the residual acetic acid. The crude residue was then purified using column chromatography eluting with petroleum ether/ethyl acetate 9:1. The title compound 6 was obtained as yellow oil (19.9 g, 83% yield). (α + β anomers): Rf = 0.18 (petroleum ether/ethyl acetate 8:2). IR: vmax (neat)/cm−1: 3300, 3032, 2937, 1590, 1310, 1042, 870. δH (400 MHz, CDCl3): 7.42–7.27 (m, 20H), 5.56–5.49 (m, 2H), 4.62–4.47 (m, 11H), 4.29–4.22 (m, 2H), 4.13–4.10 (m, 1H), 3.67–3.62 (m, 1H), 3.59–3.50 (m, 2H), 3.39–3.35 (m, 1H), 2.28–2.20 (m, 2H), 2.17–2.10 (m, 2H). δC (100.6 MHz, CDCl3): 138.1, 138.0, 137.9, 137.5, 137.3, 128.7, 128.6, 128.5, 128.48, 128.46, 128.44, 128.3, 128.1, 128.0, 127.98, 127.96, 99.4, 83.3, 82.7, 79.83, 79.8, 79.0, 78.8, 73.2, 72.1, 71.9, 71.8, 41.8, 39.2. HRMS (ESI): calculated for [M + Na]+, C19H22NaO4: 337.1416; found: 337.1421.
2.5. Synthesis of (2R,3S)-1,3-Bis(benzyloxy)hept-6-ene-2,5-diol 7
The reaction was split in two round-bottom flasks. A solution of vinyl magnesium bromide (1M in THF, 190 mL, 190 mmol, 3.0 eq.) was added to a stirred solution of 6 (19.9 g, 63.3 mmol) in dry THF (220 mL) at 0 °C under controlled atmosphere. The reaction mixture was allowed to reach room temperature and stirred for a further 24 h. The reaction mixture was cooled to 0 °C and quenched with ammonium chloride-saturated solution (50 mL), then stirred for a further 10 min at room temperature. The solvent was then evaporated under reduced pressure and the salts formed were filtered off; water (30 mL) was added, and the product was extracted using EtOAc (3 × 100 mL). The organic extracts were dried over Na2SO4 and concentrated in vacuo. The residue was purified using column chromatography on silica gel eluting with dichloromethane/ethyl acetate 8:2 to afford the mixture of two diastereoisomers 7 as a yellow oil (19.8 g, 91% yield). (α + β anomers): Rf = 0.30 and 0.46 (dichloromethane/ethyl acetate 8:2). IR: vmax (neat)/cm−1: 3422, 3064, 3031, 2868, 1737, 1643, 1497, 1454, 1371, 1245, 1208, 1092, 923, 737, 699. δH (400 MHz, CDCl3): 7.45–7.23 (m, 20H), 5.93–5.84 (m, 2H), 5.30–5.20 (m, 2H), 5.12–5.08 (m, 2H), 4.67–4.54 (m, 8H), 4.40–4.32 (m, 2H), 4.05–3.95 (m, 2H), 3.81–3.70 (m, 2H), 3.62–3.55 (m, 4H), 1.90–1.70 (m, 4H). δC (100.6 MHz, CDCl3): 141.1, 140.8, 138.0, 137.9, 128.7, 128.6, 128.2, 128.1, 128.09, 128.06, 114.6, 114.2 78.4, 77.7, 77.4, 73.6, 72.6, 72.2, 71.8, 71.7, 71.0, 70.9, 70.6, 69.8, 37.2, 37.1. HRMS (ESI): calculated for [M + Na]+, C21H26NaO4: 365.1729; found: 365.1741.
2.6. Synthesis of (2R,3S,5R)-3-(Benzyloxy)-2-((benzyloxy)methyl-5-vinyltetrahydrofuran 8β and (2R,3S,5S)-3-(Benzyloxy)-2-((benzyloxy)methyl-5-vinyltetrahydrofuran 8α
Toluene-p-sulphonyl chloride (5.49 g, 28.8 mmol, 1.1 eq.) and KOH (5 M in H2O, 13 mL, 2.5 eq.) were sequentially added to a stirred solution of 7 (9.00 g, 26.2 mmol) in acetone (250 mL). The reaction was stirred for 52 h at 35 °C (external temperature). The reaction mixture was then diluted using water and extracted using ethyl acetate (3 × 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The two diastereoisomers were separated using column chromatography eluting with petroleum ether/diethyl ether 85:15 to afford the title compounds 8α and 8β as pale yellow oils (8α 1.93 g, 23% yield; 8β 3.03 g, 36% yield).
(2R,3S,5R)-3-(benzyloxy)-2-((benzyloxy)methyl-5-vinyltetrahydrofuran 8β: [α]D20 = +21.4 (c = 4.3 in CH2Cl2). Rf = 0.56 (petroleum ether/ethyl acetate 8:2). δH (400 MHz, CDCl3): 7.39–7.21 (m, 10H), 6.01–5.93 (m, 1H), 5.25–5.08 (m, 2H), 4.65–4.39 (m, 4H), 4.37–4.31 (m, 1H), 4.16–4.13 (m, 1H), 4.04–4.02 (m, 1H), 3.86–3.82 (m, 1H), 3.77–3.73 (m, 1H), 2.35–2.28 (m, 1H), 1.91–1.86 (m, 1H). δC (100.6 MHz, CDCl3): 139.4, 138.5, 138.4, 128.5, 128.4, 127.9, 127.6, 127.5, 116.0, 81.5, 79.4, 79.2, 73.6, 71.3, 69.3, 38.5. HRMS (ESI): calculated for [M + H]+, C21H25O3: 325.1804; found: 325.1808.
(2R,3S,5S)-3-(benzyloxy)-2-((benzyloxy)methyl-5-vinyltetrahydrofuran 8α: [α]D20 = +34.0 (c = 6.0 in CH2Cl2). Rf = 0.63 (petroleum ether/ethyl acetate 8:2). IR: vmax (neat)/cm−1: 2864, 1454, 1094, 925, 787, 697. δH (400 MHz, CDCl3): 7.39–7.21 (m, 10H), 5.91–5.81 (m, 1H), 5.30–5.25 (m, 1H), 5.13–5.10 (m, 1H), 4.65–4.46 (m, 5H), 4.25–4.19 (m, 2H), 3.79 (dd, J1 = 10.0, J2 = 5.6, 1H), 3.72 (dd, J1 = 9.6, J2 = 6.4, 1H), 2.32–2.26 (m, 1H), 1.78–1.71 (m, 1H). δC (100.6 MHz, CDCl3): 138.33, 138.29, 138.2, 128.51, 128.46, 127.8, 127.74, 127.7, 116.6, 83.7, 81.5, 80.0, 77.4, 73.5, 71.2, 38.8. HRMS (ESI): calculated for [M + H]+, C21H25O3: 325.1804; found: 325.1810.
2.7. Synthesis of 1-((2R,4S,5R)-4-(Benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)ethane-1,2-diol (9a/b)
N-methylmorpholine-N-oxide (540 mg, 4.60 mmol, 1.5 eq.) and OsO4 (78 mg, 0.31 mmol, 0.1 eq.) were sequentially added to a solution of 8β (1.00 g, 3.08 mmol) in THF/H2O (1:1, 80 mL). After stirring at room temperature for 3 h, the reaction mixture was quenched using Na2S2O5/NaHSO3 (765 mg, 1.3 eq.) and stirred 1 h at the same temperature. The reaction was then extracted using ethyl acetate (3 × 50 mL). The combined organic extracts were washed with 1 N HCl (1 × 50 mL), followed by H2O (1 × 50 mL) and brine (1 × 50 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo to afford 9a/b as a mixture of two diastereoisomers (dr 80:20) as a brown oil. This product did not require any further purification for the next step (1.10 g, 99% yield). Rf = 0.24 (dichloromethane/methanol 9:1). IR: vmax (neat)/cm−1: 3384, 2936, 1445, 1064. δH (400 MHz, CDCl3): 7.42–7.22 (m, 20H), 4.69–4.51 (m, 6H), 4.42–4.39 (m, 2H), 4.27–4.11 (m, 4H), 3.99–3.97 (m, 2H), 3.91–3.89 (m, 2H), 3.79–3.61 (m, 8H), 2.29–2.20 (m, 2H), 2.14–2.08 (m, 2H). δC (100.6 MHz, CDCl3): 138.1, 137.7, 128.6, 128.54, 128.52, 128.0, 127.93, 127.9, 127.8, 127.7, 81.5, 80.9, 79.5, 78.8, 78.73, 78.67, 78.5, 73.6, 73.1, 72.8, 71.5, 71.4, 68.9, 68.8, 64.9, 64.0, 33.7, 32.0. HRMS (ESI): calculated for [M + Na]+, C21H26NaO5: 381.1678; found: 381.1681.
2.8. Synthesis of (R)-1-((2R,4S,5R)-4-(Benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)ethane-1,2-diyl Diacetate 10a and (S)-1-((2R,4S,5R)-4-(Benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)ethane-1,2-diyl Diacetate 10b
Acetic anhydride (6.2 mL, 65.0 mmol, 18.3 eq.), pyridine (3.1 mL, 38.5 mmol, 10.7 eq.), and a catalytic amount of N,N-dimethylaminopyridine (22 mg, 0.18 mmol, 0.05 eq.) were sequentially added to a stirred solution of 9a/b (1.30 g, 3.60 mmol) in CH2Cl2 (40 mL). The reaction was stirred at room temperature for 2 h, then diluted using CH2Cl2 and washed using HCl 10% (2 × 30 mL), followed by NaHCO3-saturated solution (2 × 30 mL). The organic phase was dried over Na2SO4 and then concentrated in vacuo. The reaction afforded a mixture of two diastereoisomers that were separated using column chromatography eluting with petroleum ether/diethyl ether 8:2 to afford 10a and 10b as yellow oils (10a 1.01 g, 60% yield; 10b 0.25 g, 20% yield).
(R)-1-((2R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)ethane-1,2-diyl diacetate 10a: [α]D20 = +26.6 (c = 6.17 in CH2Cl2). Rf = 0.76 (petroleum ether/ethyl acetate 6:4). IR: vmax (neat)/cm−1: 1748, 1224, 793. δH (400 MHz, CDCl3): 7.36–7.28 (m, 10H, Ar), 5.18 (ddd, J1 = 6.4, J2 = 3.6, J3 = 2.8, 1H, H-8), 4.61–4.50 (m, 4H, H-6, and H-7), 4.36 (d, J = 12, 1H, H-9), 4.18–4.01 (m, 4H, H-3, H-1, H-9′, and H-4), 3.78 (dd, J1 = 10, J2 = 4.8, 1H, H-5), 3.68 (dd, J1 = 10, J2 = 6.4, 1H, H-5′), 2.20–2.04 (m, 2H, H-2, and H-2′), 2.07 (s, 3H, H-11), 2.05 (s, 3H, H-10). δC (100.6 MHz, CDCl3): 171.0, 170.4, 138.3, 138.1, 128.5, 127.9, 127.82, 127.8, 127.76, 127.72, 82.3, 78.3, 76.2, 73.6, 73.4, 71.3, 68.9, 63.0, 33.7, 21.2, 21.0. HRMS (ESI): calculated for [M + Na]+, C25H30NaO7: 465.1889; found: 465.1883.
(S)-1-((2R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)ethane-1,2-diyl diacetate 10b: [α]D20 = +21.4 (c = 5.43 in CH2Cl2). Rf = 0.68 (petroleum ether/ethyl acetate 6:4). IR: vmax (neat)/cm−1: 1748, 1224, 793. δH (400 MHz, CDCl3): 7.34–7.27 (m, 10H, Ar), 5.24 (ddd, J1 = 6.4, J2 = 3.6, J3 = 2.8, 1H, H-8), 4.62–4.52 (m, 4H, H-6, and H-7), 4.42 (m, 1H, H-9), 4.37–4.33 (m, 1H, H-3), 4.18–4.10 (m, 2H, H-1, and H-9′), 4.04–4.00 (m, 1H, H-4), 3.80 (dd, J1 = 10, J2 = 4.8, 1H, H-5), 3.70 (dd, J1 = 10, J2 = 6.4, 1H, H-5′), 2.20–2.14 (m, 1H, H-2), 2.04 (s, 3H, H-11), 2.02 (s, 3H, H-10), 1.94–1.89 (m, 1H, H-2′). δC (100.6 MHz, CDCl3): 170.8, 128.54, 128.5, 127.9, 127.8, 127.74 127.7, 127.52, 127.5, 81.6, 78.4, 76.3, 73.6, 72.5, 71.6, 69.0, 63.6, 33.7, 21.2, 20.9. HRMS (ESI): calculated for [M + Na]+, C25H30NaO7: 465.1889; found: 465.1891.
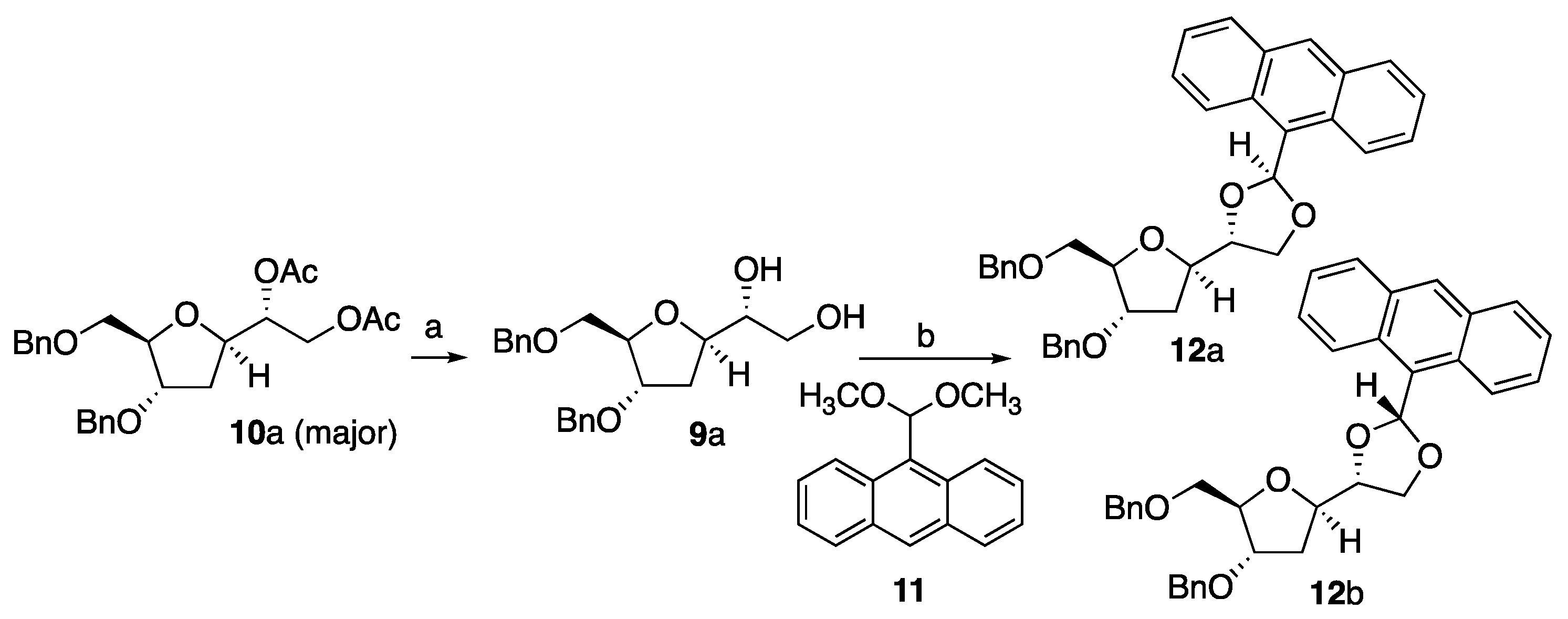
2.9. Synthesis of (2S,4R)-2-(Anthracen-9-yl)-4-((2R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)-1,3-dioxolane 12a and (2R,4R)-2-(Anthracen-9-yl)-4-((2R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)-1,3-dioxolane 12b
Anthraldehyde dimethyl acetal (59 mg, 0.23 mmol, 1.25 eq.) and p-toluensulfonic acid (0.8 mg, 2 mol%) were added to a stirred solution of 9a (67 mg, 0.19 mmol) in CH3CN (1.7 mL). The reaction was stirred at room temperature 48 h; then, it was neutralized using Et3N and the solvent was evaporated. The crude material was purified using column chromatography on silica gel eluting using petroleum ether/ethyl acetate 9:1 to afford the title compound 12a and 12b (dr 78:22) as a yellow oil (18 mg, 18%) and as individual compounds. (4R)-2-(anthracen-9-yl)-4-((2R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-yl)-1,3-dioxolane.
12a: [α]D25 = +8.6 (c = 1.4 in CH2Cl2). Rf = 0.44 (petroleum ether/ethyl acetate 8:2). IR: vmax (neat)/cm−1: 3012, 1592, 1220, 780. δH (400 MHz, CDCl3): 8.59–8.52 (m, 2H), 8.44 (s, 1H), 7.97–7.92 (m, 2H), 7.60–7.36 (m, 5H), 7.32–7.11 (m, 9H), 7.05 (s, 1H), 4.61–4.50 (m, 4H), 4.42–4.32 (m, 3H), 4.32–4.23 (m, 1H), 4.23–4.11 (m, 2H), 3.84–3.70 (m, 2H), 2.20–2.15 (m, 2H). δC (100.6 MHz, CDCl3): 138.4, 138.3, 131.6, 131.1, 130.6, 129.2, 128.5, 128.0, 127.8, 127.6, 126.3, 125.0, 124.7, 101.8, 82.3, 79.34, 79.28, 79.0, 73.7, 71.3, 69.2, 68.5, 35.1. HRMS (ESI): calculated for [M + H]+, C36H35O5: 547.2484; found: 547.2491.
12b: [α]D25 = −20.0 (c = 0.4 in CH2Cl2). Rf = 0.36 (petroleum ether/ethyl acetate 8:2). IR: vmax (neat)/cm−1:3012, 1592, 1220, 780. δH (400 MHz, CDCl3): 8.53–8.40 (m, 3H), 8.10–7.91 (m, 2H), 7.52–7.40 (m, 4H), 7.40–7.17 (m, 10H), 7.15 (s, 1H), 4.73–4.50 (m, 4H), 4.42–4.17 (m, 4H), 4.20–4.10 (m, 2H), 3.90–3.71 (m, 2H), 2.29–2.15 (m, 2H). δC (100.6 MHz, CDCl3): 138.4, 138.2, 131.6, 131.0, 130.5, 129.3, 128.6, 128.0, 127.8, 127.79, 127.7, 127.6, 126.4, 125.1, 124.9, 124.4 101.4, 82.3, 79.5, 79.0, 78.3, 73.7, 71.3, 70.0, 69.3, 35.1. HRMS (ESI): calculated for [M + H]+, C36H35O5: 547.2484; found: 547.2488.
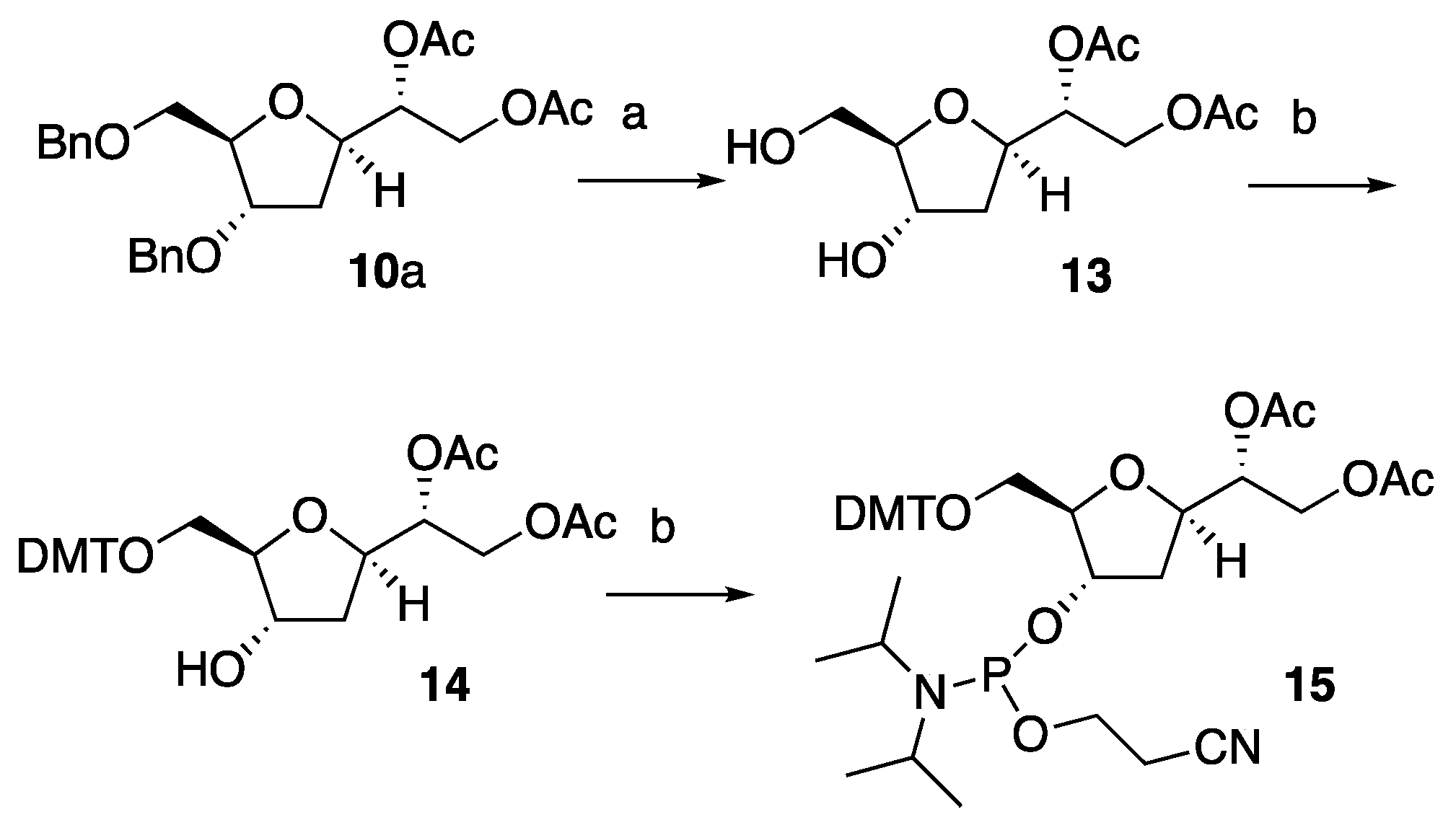
2.10. Synthesis of (R)-1-((2R,4S,5R)-4-Hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)ethane-1,2-diyl Diacetate 13
Pd/C 10% (144 mg, 1.36 mmol, 2.0 eq.) was added to a stirred solution of 10a (300 mg, 0.678 mmol) in methanol/formic acid 9:1 (24 mL). The reaction mixture was vigorously stirred at room temperature under atmospheric hydrogen pressure (balloon) for 16 h. The solution was then filtered on celite, and the solvent evaporated under reduced pressure to afford a colorless oil. This product did not require any further purification for the next step (163 mg, 92% yield). [α]D25 = +53.3 (c = 0.3 in CH2Cl2). Rf = 0.2 (dichloromethane/methanol 9:1). IR: vmax (neat)/cm−1: 3584, 2946, 1740. δH (400 MHz, CDCl3): 5.26–5.23 (m, 1H), 4.53–4.48 (m, 2H), 4.17–4.10 (m, 2H), 4.00–3.91 (m, 2H), 3.88–3.85 (m, 1H), 2.37–2.30 (m, 1H), 2.11 (s, 3H, CH3C(O)-), 2.07 (s, 3H, CH3C(O)-), 1.97–1.95 (m, 1H). δC (100.6 MHz, CDCl3): 170.9, 170.4, 77.3, 76.4, 73.5, 72.6, 62.5, 61.8, 37.2, 21.1, 21.0. HRMS (ESI): calculated for [M + Na]+, C11H18NaO7: 285.0950; found: 285.0954.
2.11. Synthesis of (R)-1-((2R,4S,5R)-5-((Bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)ethane-1,2diyl Diacetate 14
4,4’-dimethoxytrityl chloride (274 mg, 0.810 mmol, 1.3 eq.) was added to a stirred solution of 13 (163 mg g, 0.621 mmol) in dry pyridine (3.3 mL) under an inert atmosphere. The reaction was stirred at room temperature for 16 h, then quenched using a solution of chloroform/methanol 9:1 (8 mL), diluted using CH2Cl2 (5 mL) and washed using H2O (1 × 10 mL). The organic phase was dried over MgSO4, concentrated in vacuo and purified using column chromatography on silica gel eluting with petroleum ether/ethyl acetate 7:3 containing 3% of Et3N to afford 14 as a yellow oil (210 mg, 60% yield). [α]D25 = +1.8 (c = 21.0 in CH2Cl2). Rf = 0.86 (petroleum ether/ethyl acetate 1:1). IR: vmax (neat)/cm−1: 3584, 3029, 2946, 1740. δH (400 MHz, CDCl3): 7.39–7.28 (m, 4H), 7.45–7.21 (m, 5H), 6.90–6.79 (m, 4H), 5.26–5.23 (m, 1H), 4.53–4.46 (m, 2H), 4.21–4.10 (m, 1H), 3.95–3.91 (m, 1H), 3.79 (s, 6H, ArOCH3), 3.39–3.37 (m, 2H), 2.66–2.65 (m, 1H), 2.33–2.25 (m, 1H), 2.10 (s, 3H, CH3C(O)-), 2.04 (s, 3H,—CH3C(O)-), 1.97–1.91 (m, 1H). δC (100.6 MHz, CDCl3): 170.9, 170.3, 158.7, 144.7, 135.8, 130.1, 128.08, 128.04, 127.0, 113.4, 86.8, 81.7, 76.1, 73.0, 72.7, 62.9, 62.6, 55.4, 37.1, 21.2, 21.0. HRMS (ESI): calculated for [M + Na]+, C32H36NaO9: 587.2257; found: 587.2263.
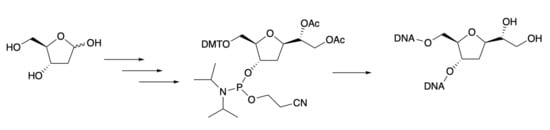
2.12. Synthesis of (1R)-1-((2R,4S,5R)-5-((Bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-(((2-cyanoethoxy)(diisopropylamino)phosphanyl)oxy)tetrahydrofuran-2-yl)ethane-1,2-diyl Diacetate 15
N,N-diisopropylethylamine (181 μL, 1.04 mmol, 2.5 eq.) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (204 μL, 0.915 mmol, 2.2 eq.) were sequentially added to a stirred solution of 14 (235 mg, 0.416 mmol) in dry CH2Cl2 (10 mL) under an inert atmosphere. The reaction was stirred at room temperature overnight, then poured into ice-cold water (15 mL) and extracted using CH2Cl2 (3 × 15 mL). The combined organic extracts were washed using water (1 × 10 mL), dried over MgSO4, and evaporated under reduced pressure. The crude residue was purified using silica gel column chromatography (petroleum ether/ethyl acetate 8:2 containing 3% Et3N) to afford 15 as a yellow oil (303 mg, 95% yield). IR: vmax (neat)/cm−1: 3029, 2946, 1740; 1380. δH (400 MHz, CDCl3): 7.49–7.57 (m, 2H), 7.38–7.19 (m, 7H), 6.91–6.77 (m, 4H), 5.11–5.07 (m, 1H), 4.61–4.53 (m, 1H), 4.47–4.05 (m, 4H), 3.79 (s, 3H), 3.78 (s, 3H), 3.65–3.12 (m, 5H), 2.58–2.51 (m, 1H), 2.45–2.20 (m, 2H), 2.15–2.12 (m, 1H), 2.08 (s, 3H), 2.06 (s, 3H), 1.97–1.91 (m, 1H), 1.21–0.90 (m, 12H). δC (100.6 MHz, CDCl3): 171.0, 170.9, 170.3, 170.1, 158.47, 158.45, 145.13, 145.1, 136.47, 136.46, 136.24, 136.21, 130.31, 130.25, 128.5, 128.4, 127.8, 126.8, 126.7, 117.9, 117.7, 113.09, 113.06, 86.23, 86.21, 83.2, 83.1, 76.01, 76.0, 73.3, 73.1, 64.3, 64.0, 62.9, 62.7, 58.7, 58.5, 58.1, 58.0, 55.33, 55.29, 43.4, 43.3, 43.2, 43.1, 36.7, 36.5, 24.74, 24.67, 24.63, 24.60, 24.55, 24.4, 24.3, 21.2, 21.1, 21.0, 20.97. HRMS (ESI): calculated for [M + Na]+, C41H53N2NaO10P: 787.3336; found: 787.3340.
2.13. Synthesis of (S)-1-((2R,4S,5R)-4-Hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)ethane-1,2-diyl Diacetate 16
Pd/C 10% (96 mg, 0.90 mmol, 2.0 eq.) was added to a stirred solution of 10b (200 mg, 0.452 mmol) in methanol/formic acid 9:1 (19 mL). The reaction mixture was vigorously stirred at room temperature under atmospheric hydrogen pressure (balloon) for 16 h. The solution was then filtered on celite, and the solvent evaporated under reduced pressure to afford a colorless oil. This product did not require any further purification for the next step (51 mg, 43% yield). [α]D25 = +40.0 (c = 0.6 in CH2Cl2). Rf = 0.2 (dichloromethane/methanol 9:1). IR: vmax (neat)/cm−1: 3584, 2946, 1740. δH (400 MHz, CDCl3): δH 5.27–5.23 (m, 1H), 4.47–4.44 (m, 1H), 4.29–4.26 (m, 1H), 4.17–4.10 (m, 2H), 3.93–3.79 (m, 3H), 2.30–2.26 (m, 1H), 2.10 (s, 3H), 2.03 (s, 3H), 1.85-1.80 (m, 1H). δC (100.6 MHz, CDCl3): 171.7, 171.2, 81.5, 77.4, 73.8, 73.7, 63.3, 62.0, 38.3, 21.4, 21.0. HRMS (ESI): calculated for [M + Na]+, C11H18NaO7: 285.0950; found: 285.0954.
2.14. Synthesis of (S)-1-((2R,4S,5R)-5-((Bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)ethane-1,2-diyl 17
4,4’-dimethoxytrityl chloride (83 mg, 0.25 mmol, 1.4 eq.) was added to a stirred solution of 16 (46 mg g, 0.175 mmol) in dry pyridine (1.0 mL) under an inert atmosphere. The reaction was stirred at room temperature for 16 h, then quenched using a solution of chloroform/methanol 9:1 (2 mL), diluted using CH2Cl2 (5 mL) and washed using H2O (1 × 5 mL). The organic phase was dried over MgSO4, concentrated in vacuo and purified using column chromatography on silica gel eluting with petroleum ether/ethyl acetate 7:3 containing 3% of Et3N to afford 17 as a yellow oil (42 mg, 43% yield). [α]D25 = +2.0 (c = 2.0 in CH2Cl2). Rf = 0.86 (petroleum ether/ethyl acetate 1:1). IR: vmax (neat)/cm−1: 3584, 3029, 2946, 1740. δH (400 MHz, CDCl3): 7.47–7.21 (m, 9H), 6.84 (d, J = 8.8, 4H), 5.26–5.24 (m, 1H), 4.42–4.38 (m, 2H), 4.26–4.15 (m, 2H), 3.89–3.88 (m, 1H), 3.79 (s, 6H), 3.42 (dd, J1 = 10, J2 = 5.2, 1H), 3.32 (dd, J1 = 10, J2 = 4.8, 1H), 2.87–2.85 (m, 1H), 2.36–2.29 (m, 1H), 2.05 (s, 3H), 2.04 (s, 3H), 1.87–1.75 (m, 1H). δC (100.6 MHz, CDCl3): 170.84, 170.82, 158.7, 144.8, 135.9, 135.8, 130.1, 128.2, 128.1, 113.4, 86.8, 81.3, 75.9, 72.7, 72.3, 63.5, 62.7, 55.4, 37.4, 21.1, 21.0. HRMS (ESI): calculated for [M + Na]+, C32H36NaO9: 587.2257; found: 587.2261.
2.15. Synthesis of (1S)-1-((2R,4S,5R)-5-((Bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-(((2-cyanoethoxy)(diisopropylamino)phosphanyl)oxy)tetrahydrofuran-2-yl)ethane-1,2diyl Diacetate 18
N,N-diisopropylethylamine (20 μL, 0.12 mmol, 2.5 eq.) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (24 μL, 0.449 mmol, 2.2 eq.) were sequentially added to a stirred solution of 17 (27 mg, 0.048 mmol) in dry CH2Cl2 (1.2 mL). The reaction was stirred at room temperature overnight under argon atmosphere, then poured into ice-cold water (5 mL) and extracted using CH2Cl2 (3 × 5 mL). The combined organic extracts were washed using water, dried over MgSO4, and evaporated under reduced pressure. The crude residue was purified using silica gel column chromatography (petroleum ether/ethyl acetate 8:2 containing 3% Et3N) to afford 18 as a yellow oil (15 mg, 41% yield). IR: vmax (neat)/cm−1: 3029, 2946, 1740; 1380. δH (400 MHz, CDCl3): 7.29–7.20 (m, 18H), 6.76–6.74 (m, 8H), 5.27–5.22 (m, 2H) 4.45–3.98 (m, 8H), 3.71 (s, 6H), 3.70 (s, 6H), 3.65–3.12 (m, 10H), 2.54–2.50 (m, 2H), 2.78–2.55 (m, 6H), 2.39–2.32 (m, 2H), 2.09 (s, 6H), 2.01 (s, 6H), 1.92–1.81 (m, 2H), 1.25–0.79 (m, 24H). δC (100.6 MHz, CDCl3): 170.84, 170.76, 170.5, 158.43, 158.4, 145.1, 136.6, 136.3, 132.5, 131.0, 130.4, 130.31, 130.28, 130.22, 128.9, 128.5, 128.4, 127.8, 126.8, 126.7, 117.8, 116.7, 113.1, 86.2, 82.1, 76.3, 76.0, 73.3, 72.7, 63.8, 63.6, 63.4, 58.2, 58.2, 55.31, 55.27, 45.42, 45.4, 43.2, 43.1, 35.9, 24.84, 24.63, 24.6, 24.59, 24.55, 24.4, 24.3, 21.2, 21.1, 21.0, 20.97. HRMS (ESI): calculated for [M + Na]+, C41H53N2NaO10P: 787.3336; found: 787.3341.
2.16. Preparation of Oligonucleotides A and B
Oligonucleotides A and B were synthesized on an AB 3400 DNA synthesizer using standard β-cyanoethyl phosphoramidite chemistry. Reagents and concentrations applied were the same as those for syntheses of natural DNA oligomers. DNA solid phase synthesis was performed on 1 μmol dABz 500 A CPG resin and 1 μmol dGiBU 500A CPG (Applied Biosystem) and using scale standard protocol. Syntheses were performed using a 1 μmol scale in trityl-on mode, according to the manufacturer’s protocol. The only change made to the usual synthesis cycle for the monomer 15 was the prolongation of the coupling time to 3 min. Coupling efficiency during the automated synthesis was estimated spectrophotometrically using the DMT cation, released during the detritylation steps. The oligomers, were removed from the support and deprotected using treatment with 35% NH3 16 h at 60 °C. The crude oligonucleotides were submitted to the protocol for PoliPak II (Glen Research) where first the DMT was removed and then the oligo purified (attached HPLC profile after Poli-Pak II treatment). After the treatment using Poli-Pak II, the sequences were submitted to RP-HPLC using a C12 Jupiter Proteo column and a gradient of 20% of B (CH3CN) in A (H2O, 0.1M TEEA, pH = 7). The products were characterized using matrix-assisted laser desorption ionization (MALDI) mass spectra using the Applied Biosystems Voyager DE-PRO spectrometer with 3-hydroxy picolinic acid matrix. Sequence A (MALDI): calculated for [M − H + Na+]− 4575.61; found: 4575.66. Sequence B (MALDI): calculated for [M − H + Na]− 4610.67; found: 4610.71.
2.17. Procedure for UV Absorption Measurements and UV-Melting Experiments
UV measurements were obtained using a JASCO V-550 UV/VIS spectrophotometer equipped with a Peltier block by using 1 cm quartz cells of both 0.5 and 1 mL internal volume (Hellma). Oligomer quantification was achieved by measuring the absorbance (l = 260 nm) at 80 °C, using the molar extinction coefficients calculated for the unstacked oligonucleotides. The molar extinction coefficients used for the calculations were A: 15.4; T: 8.8; G: 11.7; C: 7.3 m−1M−1 (for the DNA monomers). The epsilons used for the quantification of the oligonucleotide are ε260 = 152.6 m−1M−1 for sequence A (A4C2G2T6) and 165.6 m−1M−1 for sequence B (A6C2G2T4). UV quantification of the oligos provided the following values: a = 135 nmol (0.62 mg, 13% yield); b = 125 nmol (0,58 mg, 12% yield). Annealing of all the duplexes was performed by dissolving equimolar amounts of the two complementary strands in milliQ water, heating the solution at 85 °C (5 min), and then allowing to cool slowly to room temperature. Melting curves (at 260 nm) were recorded for a consecutive heating (10–85 °C)–cooling–heating protocol with a linear gradient of 0.5 °C/min.
3. Results and Discussion
A large number of synthetic approaches towards
C-nucleosides have been established to date [
23,
24]. Our group has developed a diversity-oriented strategy to provide access to a range of unnatural
C-nucleosides [
25,
26]. Taking advantage of this methodology, we set out to prepare a number of abasic nucleosides holding nonheterocyclic metal ligand templates, for example, β-diols, β-aminoalcohols, β-diamines, or β-hydroxamic acid. We set out with the synthesis of β-diol
C-nucleoside
15 (
Scheme 1) since naturally occurring nucleosides possess the β-anomeric configuration. Desired target
15 contains the protecting group required for its introduction into an oligonucleotide using solid phase synthesis. Hence, starting from commercially available 2-deoxy-D-Ribose
3, treatment with methanol in presence of catalytic AcCl generated compound
4 with a 99% yield. Subsequent exhaustive benzylation produced
5 that, in turn, was selectively deprotected on the anomeric position to provide the desired
6. Compound
6 was obtained in overall 70% yields for the steps (a)–(c) (
Scheme 1). Compound
6 was treated with an excess of vinylmagnesium bromide at room temperature to provide the corresponding ring that opened product
7 as a diastereoisomeric mixture in overall 91% isolated yields.
Diastereoisomeric mixture
7 was treated using
p-toluenesulfonyl chloride and KOH resulting in the formation of
8α/
β (
dr 1:1.5), which were successfully separated using column chromatography to obtain enantiomerically pure
8β. The
1H-NMR spectroscopy data of
8β (and therefore the stereochemistry at the anomeric position) were consistent with those already reported in the literature [
27]. The next step involved the dihydroxylation of
8β to afford diols
9a/
b. Hence, treatment of
8β with OsO
4 (10 mol%) and NMO as the terminal oxidant provided
9 in near to quantitative yield and as an inseparable mixture of two diastereoisomers. The same result was also obtained when the reaction was carried out at −78 °C. In order to increase the diastereoisomeric ratio of compound
9 and obviate to the separation of a single diastereomer, compound
8β was subjected to the condition reported by Sharpless for asymmetric dihydroxylation [
28]. Therefore,
8β was reacted in the presence of
cinchona alkaloid ligand hydroquinidine 1,4-phthalazinediyl diether (DHQD)
2PHAL [
21] (10 mol%), NMO (2.2 eq.), and OsO
4 (10 mol%). This experiment furnished
9a/
b with an inseparable mixture of two diasteroisomers. However, diols
9a/
b were then reacted with Ac
2O, pyridine and in the presence of 5% of
N,
N-dimethylaminopyridine (DMAP) to provide
10a/
b as a mixture of diastereoisomers, which, satisfactorily, could be separated using column chromatography in pure compounds
10a and
10b, respectively. Noteworthy, the preparation of compounds possessing the same scaffold as 9 and 10 has been reported using an alternative route [
29,
30,
31]. Compound
10a (major isomer) was tested for configurational stability under the standard reaction conditions adopted in oligonucleotide-automated synthesis. Hence, a solution of 7 μmol of
10a in CD
3CN (0.75 mL) was submitted to cycle reactants, including ammonia, and the progression of reaction monitored using
1H-NMR. We were delighted to observe that
10a underwent acetyl hydrolysis to provide expected
9a as a single diastereoisomer, hence proving its configurational stability under oligonucleotide synthesis conditions. The stereochemistry of the
C6–O bond of
9a and
10a was determined by converting
9a to acetal
12a and
12b and conducting n.O.e. studies on these derivatives. 9-anthraldehyde dimethyl acetal
11 has been reported as a protecting group for diols as a means to obtain crystalline structures [
21]. 9-anthraldehyde dimethyl acetal
11 (
Scheme 2) [
32] was synthesized according to the procedure reported then reacted with
9a (
Scheme 2) in MeCN under the catalysis of p-TSA to provide expected compound
12a/
b as a mixture of two diastereoisomers (
dr 78:22). Compounds
12a/
b could not be crystallized; however, it was possible, once again, to separate
12a and
12b as a single diastereoisomer using column chromatography.
With pure compounds
12a and
12b in hand, we carried out n.O.e experiments aimed at elucidating the stereochemistry of the
C4–O bond of the 1,3-dioxolane nucleus. While n.O.e. experiments carried out on
10a were unconclusive, n.O.e. run on conformationally locked
12a and
12b pointed out at the spatial orientation of the
C4–O bond in compounds compatible with an absolute (
R) stereochemistry, which can be extended to the parent compounds
9a,
10a,
12a, and
12b. In particular, upon irradiation of
C6–H in
12a, no enhancement was observed for
C1′–H but significant enhancement was observed for
C2′–H, therefore confirming a
trans relationship between
C6–H and
C1′–H; lack of enhancement of benzylic C–H upon irradiation of
C6–H was observed for compound
12a, which was in contrast to that evidenced for compound
12b. Major diastereoisomer
10a was therefore employed to obtain desired compound
15 (
Scheme 3). Firstly, hydrogenation of
10a using an excess of Pd/C (2.0 eq.) in methanol and 10% of HCOOH under an H
2 atmosphere removed the benzylic groups providing expected diol
13 in 92% isolated yields. The
5′–
O was then functionalized with a 4,4′-dimethoxytrityl group (DMT), to provide
14 at a 60% yield. In turn, compound
14 was converted to the correspondent phosphoramidite
15, which was obtained in 95% isolated yield (
Scheme 3).
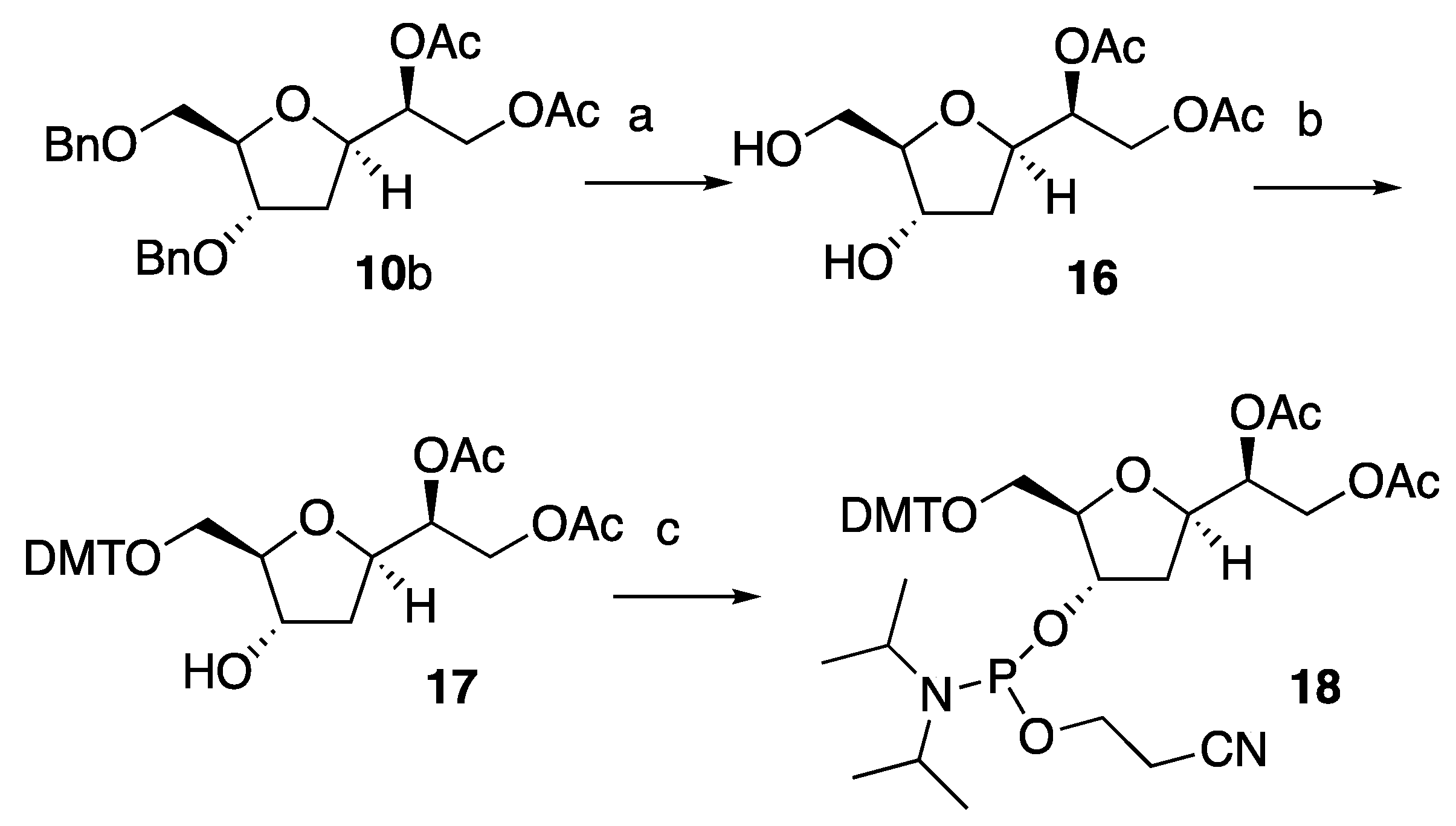
With compound
10b in hand, we repeated the synthetic route highlighted above to prepare solid phase synthesis-activated nucleoside
18 (
Scheme 4). Hence,
10b was first debenzylated under reductive conditions to generate diol
16. In turn,
16 was reacted with DMTr to provide intermediate
17 that was finally converted to the desired
18. We noted that the reaction yields for each of the steps leading to
18 were significantly lower compared to those observed for the synthesis of diastereoisomeric compound
15. These data may account for the steric hindrance provided by the C6-acetoxy group that in compounds
16 and
17 may slow the reaction of the
5′–
O and 3′–
O with their electrophilic counterparts.
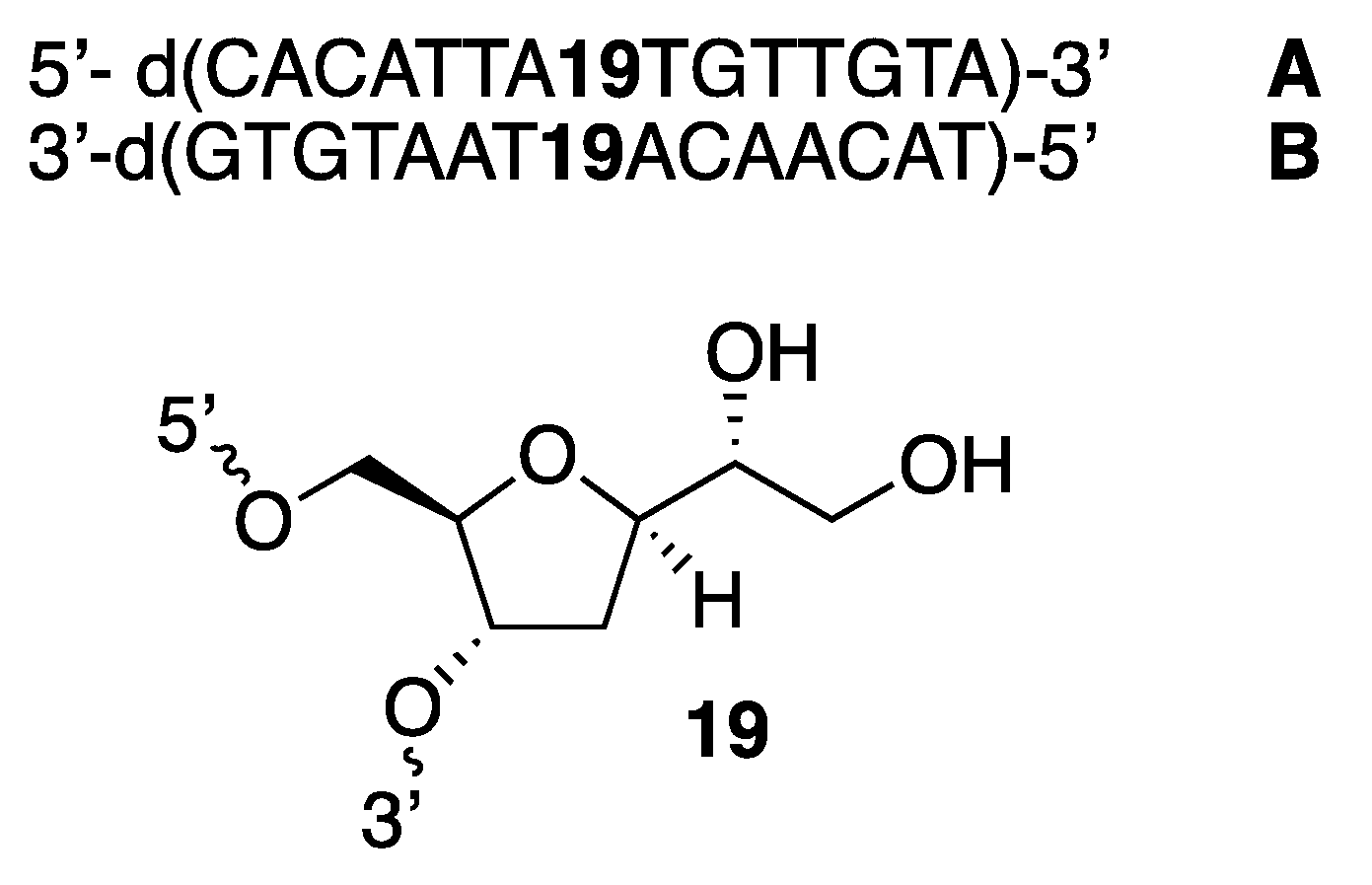
In order to evaluate the ability of abasic nucleoside
15 to be introduced on single and double strands of unnatural DNAs, compound
15 was inserted in a sequence of DNA. Hence, two strands of complementary DNAs, namely
A and
B (
Figure 2), were prepared, in which compound
15 was located in the central portion of each strand. This was achieved using standard automated DNA synthesis, demonstrating that compound
15 could efficiently be introduced in a DNA framework. This was a significant milestone, as it was shown that
15 could be used nested in a biomolecule with the prospect of becoming a catalyst upon introduction in a DNA and their subsequent deacetoxylation to become diol
19 (
Figure 2). The sequence of
A and
B was selected as reported for similar studies [
16].
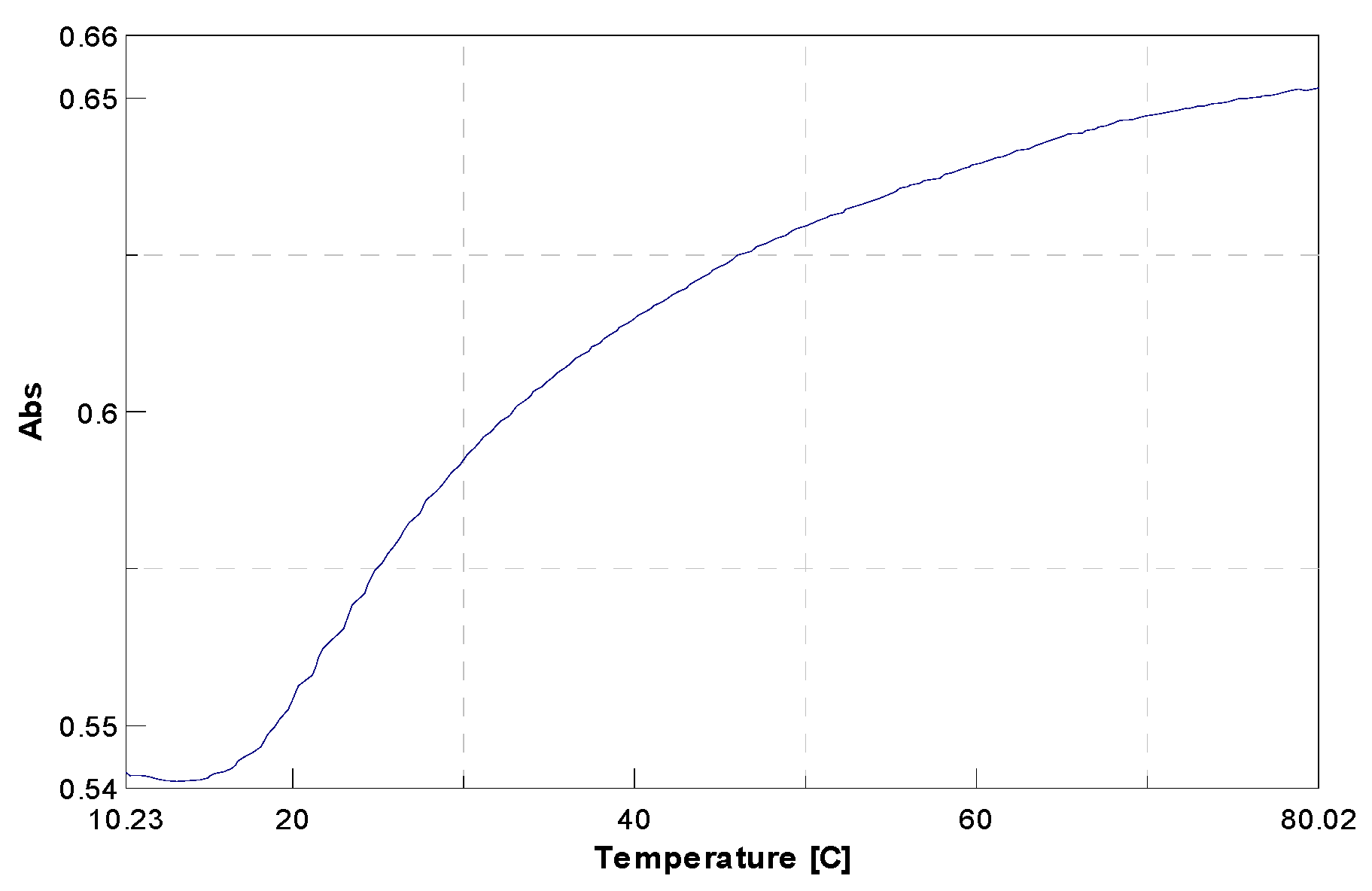
Unnatural strands A and B were then mixed and allowed to hybridize using established thermal protocols; then, the thermal stability of duplex
A/
B was recorded by carrying out UV-monitored thermal denaturation. The results obtained (
Figure 3) showed duplex
A/
B possessing a melting temperature (T
m) of 24 °C. It should be noted that in a natural-type duplex, in which the
15/
15 base pair was replaced by A-T base pair, T
m was 44.2 °C [
16]. Thus, these data show that the introduction of
15 in a natural sequence of DNA perturbed the overall stability of the duplex, resulting in a significant decrease in melting temperature (ΔT
m = 20.2 °C). The data were significant, as the lower meting temperature obtained by introducing nucleobase
15 indicated the formation of a new groove with potential for nucleophilic catalysis or for metal coordination.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






