Artificial Intelligence Algorithms for Discovering New Active Compounds Targeting TRPA1 Pain Receptors



Abstract
:1. Introduction
2. Experiments and Methods
2.1. Datasets Preparation
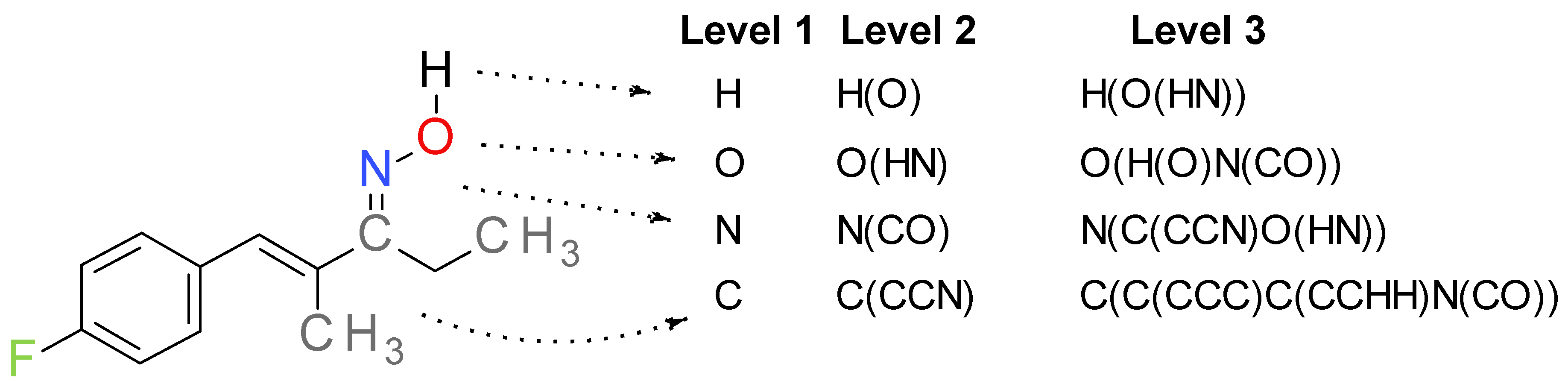
2.2. Descriptors Generation
2.3. Feedforward Neural Networks (FFNN)
2.4. Random Forest
2.5. Support Vector Machine
2.6. Performance Metrics
3. Results
3.1. TRPA1 Inhibitors and Decoys Datasets
3.2. Descriptors Generation
3.3. Machine Learning Performance Metrics
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef]
- Moparthi, L.; Kichko, T.I.; Eberhardt, M.; Högestätt, E.D.; Kjellbom, P.; Johanson, U.; Reeh, P.W.; Leffler, A.; Filipovic, M.R.; Zygmunt, P.M. Human TRPA1 is a heat sensor displaying intrinsic U-shaped thermosensitivity. Sci. Rep. 2016, 6, 28763. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Jordt, S.-E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Orengo, L.; Dhaka, A.; Heuermann, R.J.; Young, T.J.; Montana, M.C.; Cavanaugh, E.J.; Kim, D.; Story, G.M. Cutaneous Nociception Evoked by 15-delta PGJ2 via Activation of Ion Channel TRPA1. Mol. Pain 2008, 4, 1730–1744. [Google Scholar] [CrossRef] [Green Version]
- Sisignano, M.; Park, C.-K.; Angioni, C.; Zhang, D.D.; von Hehn, C.; Cobos, E.J.; Ghasemlou, N.; Xu, Z.-Z.; Kumaran, V.; Lu, R.; et al. 5,6-EET Is Released upon Neuronal Activity and Induces Mechanical Pain Hypersensitivity via TRPA1 on Central Afferent Terminals. J. Neurosci. 2012, 32, 6364–6372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevisan, G.; Benemei, S.; Materazzi, S.; De Logu, F.; De Siena, G.; Fusi, C.; Fortes Rossato, M.; Coppi, E.; Marone, I.M.; Ferreira, J.; et al. TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain 2016, 139, 1361–1377. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, G.; Hoffmeister, C.; Rossato, M.F.; Oliveira, S.M.; Silva, M.A.; Silva, C.R.; Fusi, C.; Tonello, R.; Minocci, D.; Guerra, G.P.; et al. TRPA1 receptor stimulation by hydrogen peroxide is critical to trigger hyperalgesia and inflammation in a model of acute gout. Free Radic. Biol. Med. 2014, 72, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, C.; Zhu, S.; Zhou, N. TRPA1: The Central Molecule for Chemical Sensing in Pain Pathway? J. Neurosci. 2008, 28, 1019–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivisto, A.; Jalava, N.; Bratty, R.; Pertovaara, A. TRPA1 Antagonists for Pain Relief. Pharmaceuticals 2018, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-X.; Zhang, Y. Emerging drug targets for pain treatment. Eur. J. Pharmacol. 2012, 681, 1–5. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Akhoon, B.A.; Tiwari, H.; Nargotra, A. In Silico Drug Design Methods for Drug Repurposing. In In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 47–84. ISBN 978-0-12-816125-8. [Google Scholar]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahikkala, T.; Airola, A.; Pietila, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug-target interaction predictions. Brief. Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, D.J.; Manallack, D.T.; Tetko, I. V Data modelling with neural networks: Advantages and limitations. J. Comput.-Aided Mol. Des. 1997, 11, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [Green Version]
- Kinnings, S.L.; Jackson, R.M. ReverseScreen3D: A Structure-Based Ligand Matching Method to Identify Protein Targets. J. Chem. Inf. Modeling 2011, 51, 624–634. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, L.; Yin, Z.; Lin, J. Improving chemical similarity ensemble approach in target prediction. J. Cheminformatics 2016, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yul, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J.; et al. TarFisDock: A web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006, 34, W219–W224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonov, D.A.; Druzhilovskiy, D.S.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Dmitriev, A.V.; Pogodin, P.V.; Poroikov, V.V. Computer-aided prediction of biological activity spectra for chemical compounds: Opportunities and limitation. Biomed. Chem. Res. Methods 2018, 1, e00004. [Google Scholar] [CrossRef] [Green Version]
- Geronikaki, A.; Lagunin, A.; Poroikov, V.; Filimonov, D.; Hadjipavlou-Litina, D.; Vicini, P. Computer aided prediction of biological activity spectra: Evaluating versus known and predicting of new activities for thiazole derivatives. SAR QSAR Environ. Res. 2002, 13, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, Q.-J.; Pan, J.; Yang, Y.; Wu, X.-P. A prediction model for blood–brain barrier permeation and analysis on its parameter biologically. Comput. Methods Programs Biomed. 2009, 95, 280–287. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. Support vector machines for drug discovery. Expert Opin. Drug Discov. 2014, 9, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lind, A.P.; Anderson, P.C. Predicting drug activity against cancer cells by random forest models based on minimal genomic information and chemical properties. PLoS ONE 2019, 14, e0219774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltarollo, V.G.; Kronenberger, T.; Espinoza, G.Z.; Oliveira, P.R.; Honorio, K.M. Advances with support vector machines for novel drug discovery. Expert Opin. Drug Discov. 2019, 14, 23–33. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Modeling 2015, 55, 460–473. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Keras-team. GitHub. 2005. Available online: https://github.com/keras-team/keras (accessed on 10 June 2020).
- Hinton, G.E.; Srivastava, N.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R.R. Improving neural networks by preventing co-adaptation of feature detectors. arXiv Preprint 2012, arXiv:1207.0580. Available online: https://arxiv.org/pdf/1207.0580.pdf (accessed on 11 June 2020).
- Golik, P.; Doetsch, P.; Ney, H. Cross-Entropy vs. Squared Error Training: A Theoretical and Experimental Comparison. In Proceedings of the 14th Annual Conference of the International Speech Communication Association, Lyon, France, 25–29 August 2013; pp. 1756–1760. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Kumar, R.; Indrayan, A. Receiver operating characteristic (ROC) curve for medical researchers. Indian Pediatr. 2011, 48, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Saponaro, G.; Szallasi, A. Transient receptor potential ankyrin 1 (TRPA1) antagonists. Pharm. Pat. Anal. 2015, 4, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Mihai, D.P.; Nitulescu, G.M.; Ion, G.N.D.; Ciotu, C.I.; Chirita, C.; Negres, S. Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis. Pharmaceutics 2019, 11, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A. Robustness of Biological Activity Spectra Predicting by Computer Program PASS for Noncongeneric Sets of Chemical Compounds. J. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Algorithm | TPR (%) | TNR (%) | ACC (%) | bACC (%) | FPR (%) | NPV (%) | Mean ROC AUC |
|---|---|---|---|---|---|---|---|
| RF | 100 | 96 | 99 | 98 | 4 | 100 | 0.9936 |
| SVM | 92 | 84 | 90 | 88 | 16 | 77.78 | 0.9354 |
| FFNN | 90.67 | 80 | 88 | 85.33 | 20 | 74.07 | 0.9354 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihai, D.P.; Trif, C.; Stancov, G.; Radulescu, D.; Nitulescu, G.M. Artificial Intelligence Algorithms for Discovering New Active Compounds Targeting TRPA1 Pain Receptors. AI 2020, 1, 276-285. https://doi.org/10.3390/ai1020018
Mihai DP, Trif C, Stancov G, Radulescu D, Nitulescu GM. Artificial Intelligence Algorithms for Discovering New Active Compounds Targeting TRPA1 Pain Receptors. AI. 2020; 1(2):276-285. https://doi.org/10.3390/ai1020018
Chicago/Turabian StyleMihai, Dragos Paul, Cosmin Trif, Gheorghe Stancov, Denise Radulescu, and George Mihai Nitulescu. 2020. "Artificial Intelligence Algorithms for Discovering New Active Compounds Targeting TRPA1 Pain Receptors" AI 1, no. 2: 276-285. https://doi.org/10.3390/ai1020018
APA StyleMihai, D. P., Trif, C., Stancov, G., Radulescu, D., & Nitulescu, G. M. (2020). Artificial Intelligence Algorithms for Discovering New Active Compounds Targeting TRPA1 Pain Receptors. AI, 1(2), 276-285. https://doi.org/10.3390/ai1020018