
Effects of Chemical Short-Range Order and Temperature on Basic Structure Parameters and Stacking Fault Energies in Multi-Principal Element Alloys


Abstract
:1. Introduction
2. Materials and Methods



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPEA | Methods | ||||
|---|---|---|---|---|---|
| CoCrNi | Exp | 3.565 [60] | |||
| DFT | 3.305 [61] | 278.8 [62] | 188.8 [62] | 183.0 [62] | |
| EAM | 3.55644 | 251.644 | 178.246 | 95.134 | |
| DFT | 3.26 | 261.51 | 181.48 | 61.72 | |
| MoNbTa | EAM | 3.243 | 307.17 | 159.46 | 77.51 |
| HfMoNbTaTi | Exp | 3.305 [13,14] | |||
| DFT [45] | 3.305 | 209.95 | 133.92 | 39.68 | |
| EAM [45] | 3.31 | 201.42 | 146.64 | 82 | |
| HfNbTaTiZr | Exp | 3.304 [15] | 172 [63] | 116 [63] | 28 [63] |
| DFT [64] | 3.457 | 160.2 | 124.4 | 62.4 | |
| EAM [45] | 3.405 | 149.63 | 111.83 | 64.67 |
3. Results and Discussions
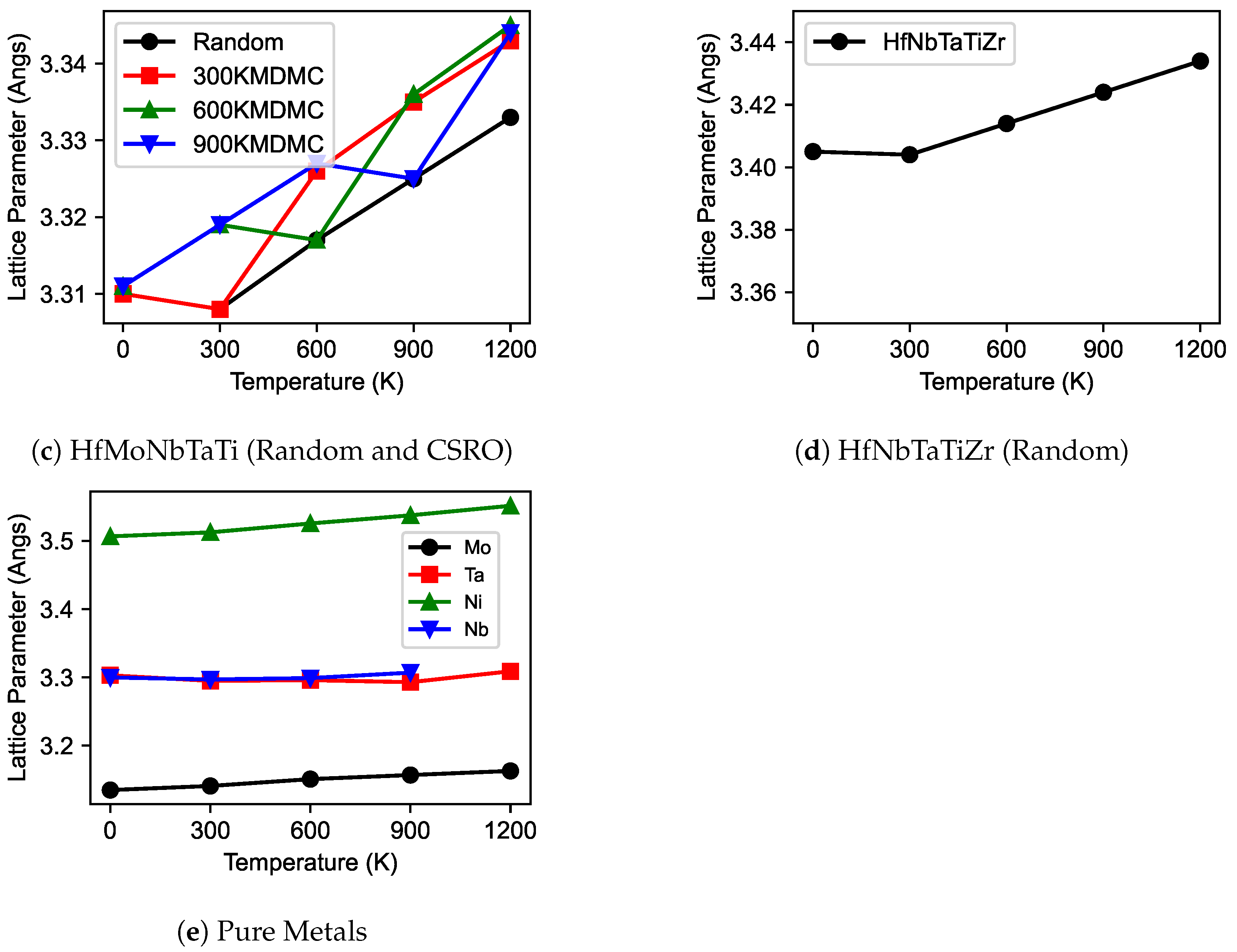
3.1. Lattice Parameters
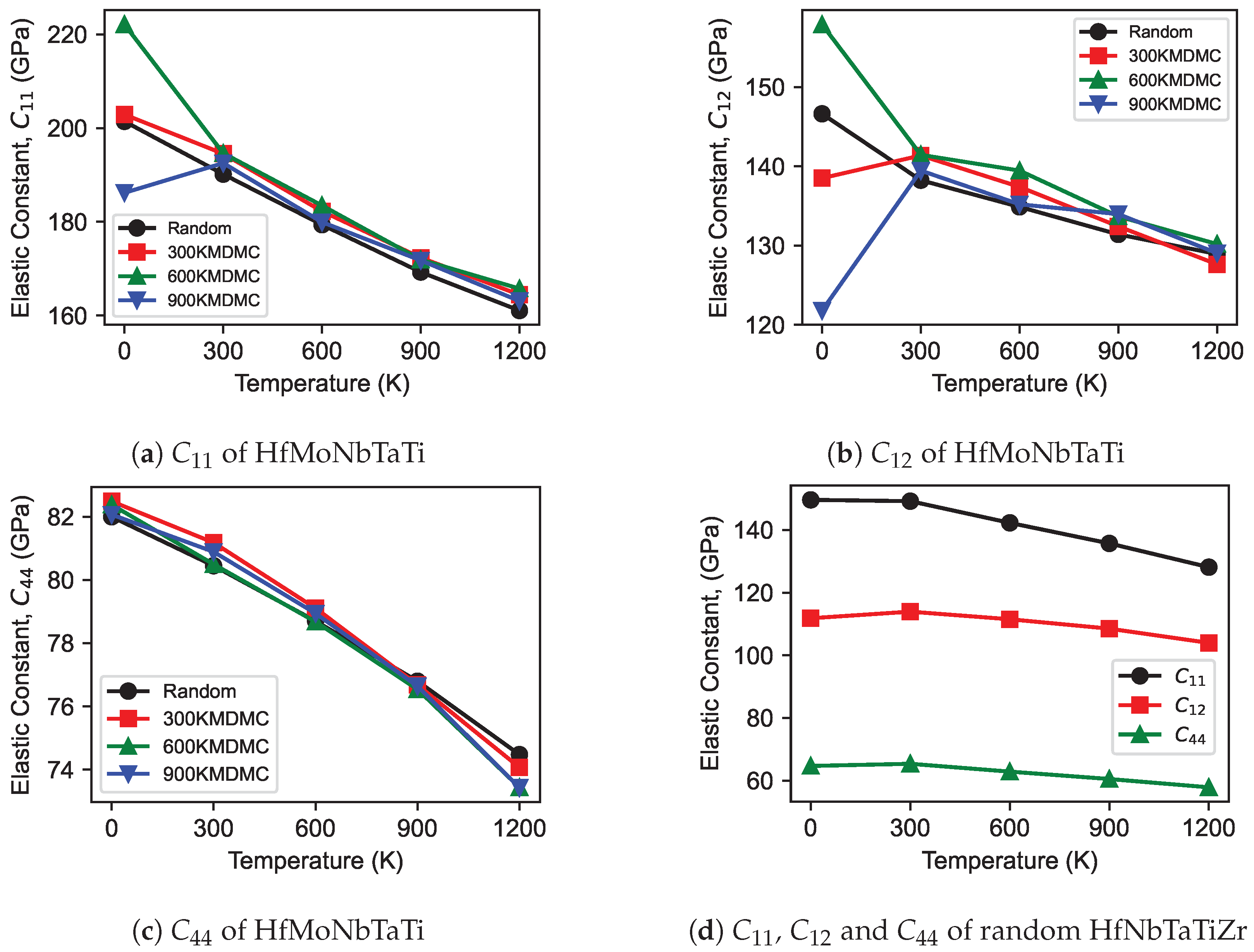
3.2. Elastic Constants
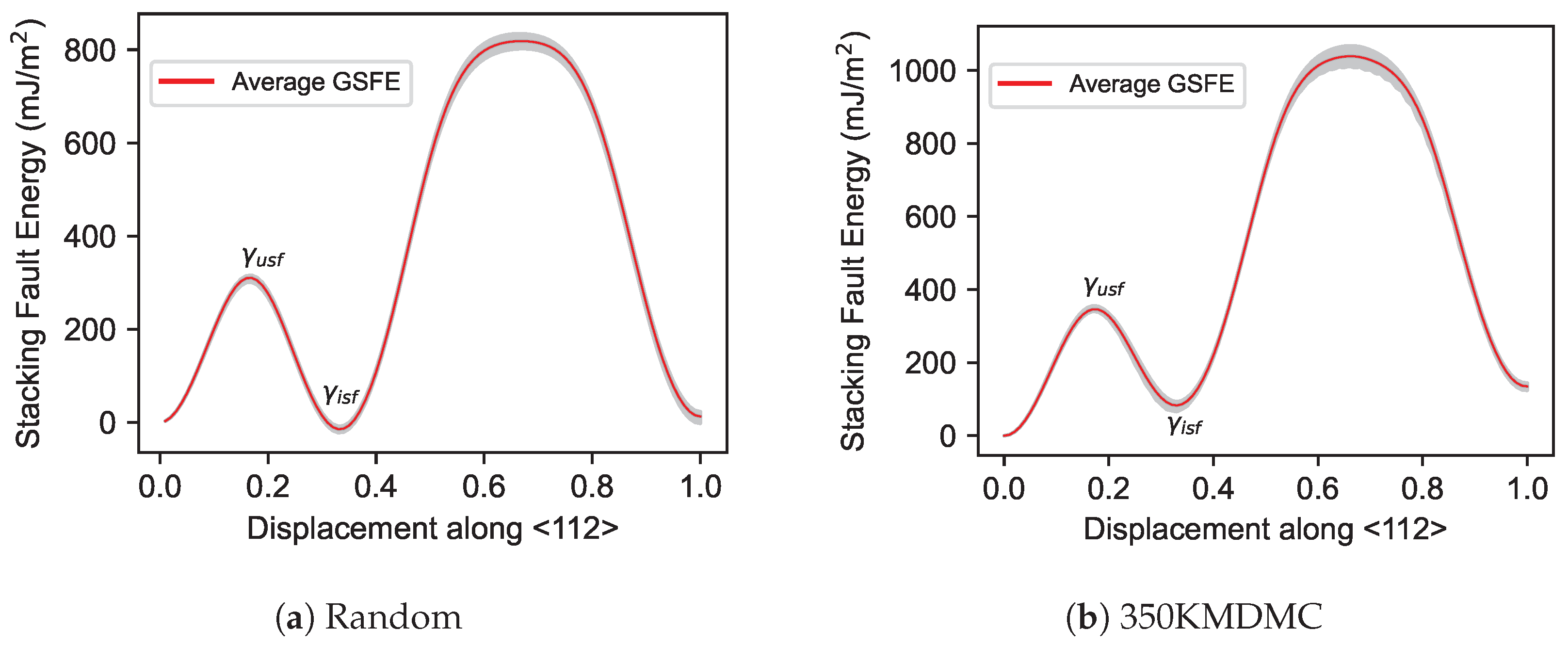
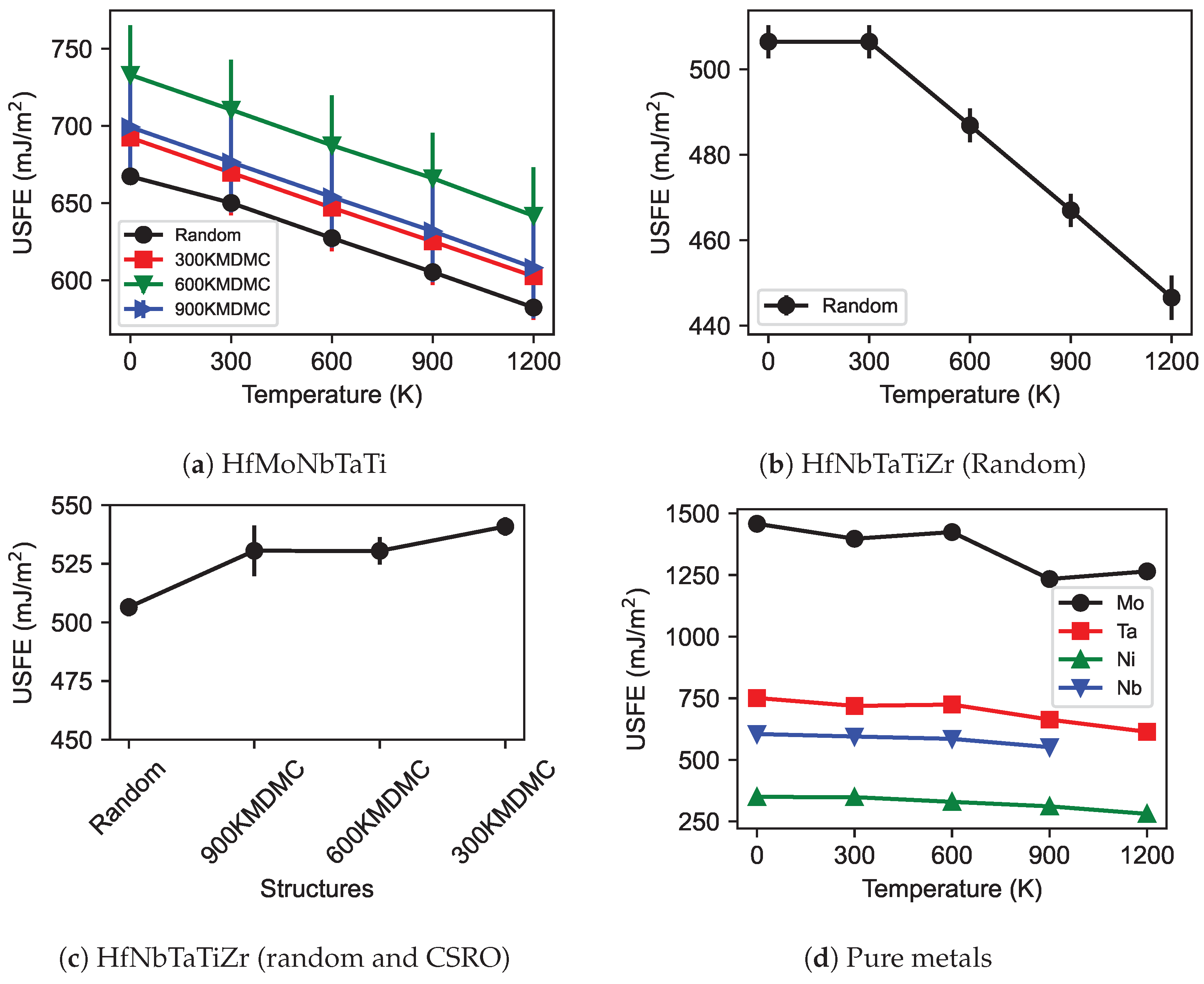
3.3. Generalized Stacking Fault Energy (GSFE)
4. Conclusions
- The investigation revealed that an increase in temperature leads to an expansion in the lattice parameter across all studied MPEAs and pure metals. For CoCrNi and MoNbTa, the lattice parameters showed minimal variation between CSRO and random structures.
- Consistent with expectations, a temperature rise resulted in a decrease in the stiffness of both MPEAs and pure metals, thus reducing their elastic constants. Notably, CSRO structures demonstrated greater stiffness relative to random structured MPEAs. The trend in elastic constants for both random and CSRO structures was similar, with a decrease observed as the temperature increased. Additionally, CSRO structures manifested higher GSFEs than random structures.
- Understanding the role of CSRO and temperature in determining material properties allows for the fine-tuning of alloys to achieve desired mechanical properties, including ductility, strength, and resistance to deformation mechanisms like slip and twinning. It supports the development of more accurate models for predicting material behavior, facilitating the exploration of new alloys and composite materials. This work lays a foundational basis for further investigations into the mechanical characteristics of various alloys, encouraging a more in-depth exploration of dislocation dynamics and the effects of elemental concentrations on plastic deformation behavior.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Structural Parameters and GSFE of MoNbTa Calculated via DFT
References
- Cantor, B.; Chang, I.T.H.; Knight, P.; Vincent, A.J.B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375-377, 213–218. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Ma, E. Unusual dislocation behavior in high-entropy alloys. Scr. Mater. 2020, 181, 127–133. [Google Scholar] [CrossRef]
- Nöhring, W.G.; Curtin, W.A. Design using randomness: A new dimension for metallurgy. Scr. Mater. 2020, 187, 210–215. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, S.; Ritchie, R.O.; Meyers, M.A. Mechanical properties of high-entropy alloys with emphasis on face-centered cubic alloys. Prog. Mater. Sci. 2019, 102, 296–345. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Thurston, K.V.S.; Bei, H.; Wu, Z.; George, E.P.; Ritchie, R.O. Exceptional damage-tolerance of a medium-entropy alloy CrCoNi at cryogenic temperatures. Nat. Commun. 2016, 7, 10602. [Google Scholar] [CrossRef]
- Zeng, Z.; Xiang, M.; Zhang, D.; Shi, J.; Wang, W.; Tang, X.; Tang, W.; Wang, Y.; Ma, X.; Chen, Z.; et al. Mechanical properties of Cantor alloys driven by additional elements: A review. J. Mater. Res. Technol. 2021, 15, 1920–1934. [Google Scholar] [CrossRef]
- Senkov, O.N.; Miracle, D.B.; Chaput, K.J.; Couzinie, J.P. Development and exploration of refractory high entropy alloys—A review. J. Mater. Res. 2018, 33, 3092–3128. [Google Scholar] [CrossRef]
- Senkov, O.N.; Wilks, G.B.; Miracle, D.B.; Chuang, C.P.; Liaw, P.K. Refractory high-entropy alloys. Intermetallics 2010, 18, 1758–1765. [Google Scholar] [CrossRef]
- Wadsworth, J.; Nieh, T.G.; Stephens, J.J. Recent advances in aerospace refractory metal alloys. Int. Mater. Rev. 1988, 33, 131–150. [Google Scholar] [CrossRef]
- Tseng, K.K.; Juan, C.C.; Tso, S.; Chen, H.C.; Tsai, C.W.; Yeh, J.W. Effects of Mo, Nb, Ta, Ti, and Zr on Mechanical Properties of Equiatomic Hf-Mo-Nb-Ta-Ti-Zr Alloys. Entropy 2019, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Bian, H.; Aoyagi, K.; Hayasaka, Y.; Yamanaka, K.; Chiba, A. Synergetic strengthening in HfMoNbTaTi refractory high-entropy alloy via disordered nanoscale phase and semicoherent refractory particle. Mater. Des. 2021, 212, 110248. [Google Scholar] [CrossRef]
- Senkov, O.N.; Scott, J.M.; Senkova, S.V.; Miracle, D.B.; Woodward, C.F. Microstructure and room temperature properties of a high-entropy TaNbHfZrTi alloy. J. Alloys Compd. 2011, 509, 6043–6048. [Google Scholar] [CrossRef]
- Senkov, O.N.; Scott, J.M.; Senkova, S.V.; Meisenkothen, F.; Miracle, D.B.; Woodward, C.F. Microstructure and elevated temperature properties of a refractory TaNbHfZrTi alloy. Mater. Sci. Eng. A 2012, 47, 4062–4074. [Google Scholar] [CrossRef]
- Couzinié, J.P.; Lilensten, L.; Champion, Y.; Dirras, G.; Perrière, L.; Guillot, I. On the room temperature deformation mechanisms of a TiZrHfNbTa refractory high-entropy alloy. Mater. Sci. Eng. A 2015, 645, 255–263. [Google Scholar] [CrossRef]
- An, Z.; Mao, S.; Liu, Y.; Wang, L.; Zhou, H.; Gan, B.; Zhang, Z.; Han, X. A novel HfNbTaTiV high-entropy alloy of superior mechanical properties designed on the principle of maximum lattice distortion. J. Mater. Sci. Technol. 2021, 79, 109–117. [Google Scholar] [CrossRef]
- Yasuda, H.Y.; Yamada, Y.; Cho, K.; Nagase, T. Deformation behavior of HfNbTaTiZr high entropy alloy singe crystals and polycrystals. Mater. Sci. Eng. A 2021, 809, 140983. [Google Scholar] [CrossRef]
- Juan, C.C.; Tsai, M.H.; Tsai, C.W.; Hsu, W.L.; Lin, C.M.; Chen, S.K.; Lin, S.J.; Yeh, J.W. Simultaneously increasing the strength and ductility of a refractory high-entropy alloy via grain refining. Mater. Lett. 2016, 184, 200–203. [Google Scholar] [CrossRef]
- Senkov, O.N.; Pilchak, A.L.; Semiatin, S.L. Effect of Cold Deformation and Annealing on the Microstructure and Tensile Properties of a HfNbTaTiZr Refractory High Entropy Alloy. Metall. Mater. Trans. A 2018, 49, 2876–2892. [Google Scholar] [CrossRef]
- Chen, S.Y.; Wang, L.; Li, W.D.; Tong, Y.; Tseng, K.K.; Tsai, C.W.; Yeh, J.W.; Ren, Y.; Guo, W.; Poplawsky, J.D.; et al. Peierls barrier characteristic and anomalous strain hardening provoked by dynamic-strain-aging strengthening in a body-centered-cubic high-entropy alloy. Mater. Res. Lett. 2019, 7, 475–481. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, Y.; Li, J.; Jiang, C.; Liu, B.; Liu, Y.; Liaw, P.K. Probing the phase transformation and dislocation evolution in dual-phase high-entropy alloys. Int. J. Plast. 2019, 114, 161–173. [Google Scholar] [CrossRef]
- Smith, L.T.W.; Su, Y.; Xu, S.; Hunter, A.; Beyerlein, I.J. The effect of local chemical ordering on Frank-Read source activation in a refractory multi-principal element alloy. Int. J. Plast. 2020, 134, 102850. [Google Scholar] [CrossRef]
- Hua, D.; Xia, Q.; Wang, W.; Zhou, Q.; Li, S.; Qian, D.; Shi, J.; Wang, H. Atomistic insights into the deformation mechanism of a CoCrNi medium entropy alloy under nanoindentation. Int. J. Plast. 2021, 142, 102997. [Google Scholar] [CrossRef]
- Hull, D.; Bacon, D.J. Introduction to Dislocations; Butterworth-Heinemann: Oxford, UK, 2001. [Google Scholar]
- Vitek, V. Atomic level computer modelling of crystal defects with emphasis on dislocations: Past, present and future. Prog. Mater. Sci. 2011, 56, 577–585. [Google Scholar] [CrossRef]
- Vitek, V. Structure of dislocation cores in metallic materials and its impact on their plastic behaviour. Prog. Mater. Sci. 1992, 36, 1–27. [Google Scholar] [CrossRef]
- Su, Y.; Xu, S.; Beyerlein, I.J. Density functional theory calculations of generalized stacking fault energy surfaces for eight face-centered cubic transition metals. J. Appl. Phys. 2019, 126, 105112. [Google Scholar] [CrossRef]
- Mayahi, R. An investigation concerning generalized stacking fault behavior of AlCoxCrFeNi high entropy alloys: Insights from first-principles study. J. Alloys Compd. 2020, 818, 152928. [Google Scholar] [CrossRef]
- Jarlöv, A.; Ji, W.; Zhu, Z.; Tian, Y.; Babicheva, R.; An, R.; Seet, H.L.; Nai, M.L.S.; Zhou, K. Molecular dynamics study on the strengthening mechanisms of Cr–Fe–Co–Ni high-entropy alloys based on the generalized stacking fault energy. J. Alloys Compd. 2022, 905, 164137. [Google Scholar] [CrossRef]
- Huang, S.; Luo, S.; Qin, L.; Shu, D.; Sun, B.; Lunt, A.J.G.; Korsunsky, A.M.; Mi, J. 3D local atomic structure evolution in a solidifying Al-0.4Sc dilute alloy melt revealed in operando by synchrotron X-ray total scattering and modelling. Scr. Mater. 2022, 211, 114484. [Google Scholar] [CrossRef]
- Choudhuri, D.; Majumdar, B.S.; Wilkinson, H. Investigation of in-liquid ordering mediated transformations in Al-Sc via ab initio molecular dynamics and unsupervised learning. Phys. Rev. Mater. 2022, 6, 103406. [Google Scholar] [CrossRef]
- Li, X.G.; Chen, C.; Zheng, H.; Zuo, Y.; Ong, S.P. Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy. npj Comput. Mater. 2020, 6, 70. [Google Scholar] [CrossRef]
- Zhao, L.; Zong, H.; Ding, X.; Lookman, T. Anomalous dislocation core structure in shock compressed bcc high-entropy alloys. Acta Mater. 2021, 209, 116801. [Google Scholar] [CrossRef]
- Yin, S.; Zuo, Y.; Abu-Odeh, A.; Zheng, H.; Li, X.G.; Ding, J.; Ong, S.P.; Asta, M.; Ritchie, R.O. Atomistic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short-range order. Nat. Commun. 2021, 12, 4873. [Google Scholar] [CrossRef]
- Zheng, H.; Fey, L.T.W.; Li, X.G.; Hu, Y.J.; Qi, L.; Chen, C.; Xu, S.; Beyerlein, I.J.; Ong, S.P. Multi-scale Investigation of Chemical Short-Range Order and Dislocation Glide in the MoNbTi and TaNbTi Refractory Multi-Principal Element Alloys. npj Comput. Mater. 2023, 9, 89. [Google Scholar] [CrossRef]
- An, X.H.; Wu, S.D.; Wang, Z.G.; Zhang, Z.F. Significance of stacking fault energy in bulk nanostructured materials: Insights from Cu and its binary alloys as model systems. Prog. Mater. Sci. 2019, 101, 1–45. [Google Scholar] [CrossRef]
- Ziehl, T.J.; Morris, D.; Zhang, P. Detection and impact of short-range order in medium/high-entropy alloys. iScience 2023, 26, 106209. [Google Scholar] [CrossRef]
- Birbilis, N.; Choudhary, S.; Scully, J.R.; Taheri, M.L. A perspective on corrosion of multi-principal element alloys. npj Mater. Degrad. 2021, 5, 14. [Google Scholar] [CrossRef]
- Wang, S.D.; Liu, X.J.; Lei, Z.F.; Lin, D.Y.; Bian, F.G.; Yang, C.M.; Jiao, M.Y.; Du, Q.; Wang, H.; Wu, Y.; et al. Chemical short-range ordering and its strengthening effect in refractory high-entropy alloys. Phys. Rev. B 2021, 103, 104107. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Li, Q.J.; Sheng, H.; Ma, E. Strengthening in multi-principal element alloys with local-chemical-order roughened dislocation pathways. Nat. Commun. 2019, 10, 3563. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.H.; Wang, Y.J.; Dai, L.H. Novel atomic-scale mechanism of incipient plasticity in a chemically complex CrCoNi medium-entropy alloy associated with inhomogeneity in local chemical environment. Acta Mater. 2020, 194, 283–294. [Google Scholar] [CrossRef]
- Xu, S.; Jian, W.R.; Beyerlein, I.J. Ideal simple shear strengths of two HfNbTaTi-based quinary refractory multi-principal element alloys. APL Mater. 2022, 10, 111107. [Google Scholar] [CrossRef]
- Xu, S.; Chavoshi, S.Z.; Su, Y. On calculations of basic structural parameters in multi-principal element alloys using small atomistic models. Comput. Mater. Sci 2022, 202, 110942. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Xu, S.; Hwang, E.; Jian, W.R.; Su, Y.; Beyerlein, I.J. Atomistic calculations of the generalized stacking fault energies in two refractory multi-principal element alloys. Intermetallics 2020, 124, 106844. [Google Scholar] [CrossRef]
- Rao, S.I.; Akdim, B.; Antillon, E.; Woodward, C.; Parthasarathy, T.A.; Senkov, O.N. Modeling solution hardening in BCC refractory complex concentrated alloys: NbTiZr, Nb1.5TiZr0.5 and Nb0.5TiZr1.5. Acta Mater. 2019, 168, 222–236. [Google Scholar] [CrossRef]
- Maresca, F.; Curtin, W.A. Theory of screw dislocation strengthening in random BCC alloys from dilute to “High-Entropy” alloys. Acta Mater. 2020, 182, 144–162. [Google Scholar] [CrossRef]
- Maresca, F.; Curtin, W.A. Mechanistic origin of high strength in refractory BCC high entropy alloys up to 1900K. Acta Mater. 2020, 182, 235–249. [Google Scholar] [CrossRef]
- Yan, S.; Qi, Z.; Chen, Y.; Cao, Y.; Zhang, J.; Zheng, G.; Chen, F.; Bian, T.; Chen, G. Interlamellar boundaries govern cracking. Acta Mater. 2021, 215, 117091. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, Y.; Qi, Z.; Chen, G. Increasing high-temperature fatigue resistance of polysynthetic twinned TiAl single crystal by plastic strain delocalization. J. Mater. Sci. Technol. 2021, 93, 53–59. [Google Scholar] [CrossRef]
- Ikehata, H.; Nagasako, N.; Furuta, T.; Fukumoto, A.; Miwa, K.; Saito, T. First-principles calculations for development of low elastic modulus Ti alloys. Phys. Rev. B 2004, 70, 174113. [Google Scholar] [CrossRef]
- Jian, W.R.; Xie, Z.; Xu, S.; Su, Y.; Yao, X.; Beyerlein, I.J. Effects of lattice distortion and chemical short-range order on the mechanisms of deformation in medium entropy alloy CoCrNi. Acta Mater. 2020, 199, 352–369. [Google Scholar] [CrossRef]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commu. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Cowley, J.M. An Approximate Theory of Order in Alloys. Phys. Rev. 1950, 77, 669–675. [Google Scholar] [CrossRef]
- De Fontaine, D. The number of independent pair-correlation functions in multicomponent systems. J. Appl. Crystallogr. 1971, 4, 15–19. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Moravcik, I.; Cizek, J.; Kovacova, Z.; Nejezchlebova, J.; Kitzmantel, M.; Neubauer, E.; Kubena, I.; Hornik, V.; Dlouhy, I. Mechanical and microstructural characterization of powder metallurgy CoCrNi medium entropy alloy. Mater. Sci. Eng. A 2017, 701, 370–380. [Google Scholar] [CrossRef]
- Ye, Y.F.; Zhang, Y.H.; He, Q.F.; Zhuang, Y.; Wang, S.; Shi, S.Q.; Hu, A.; Fan, J.; Yang, Y. Atomic-scale distorted lattice in chemically disordered equimolar complex alloys. Acta Mater. 2018, 150, 182–194. [Google Scholar] [CrossRef]
- Ge, H.; Song, H.; Shen, J.; Tian, F. Effect of alloying on the thermal-elastic properties of 3d high-entropy alloys. Mater. Chem. Phys. 2018, 210, 320–326. [Google Scholar] [CrossRef]
- Dirras, G.; Lilensten, L.; Djemia, P.; Laurent-Brocq, M.; Tingaud, D.; Couzinié, J.P.; Perrière, L.; Chauveau, T.; Guillot, I. Elastic and plastic properties of as-cast equimolar TiHfZrTaNb high-entropy alloy. Mater. Sci. Eng. A 2016, 654, 30–38. [Google Scholar] [CrossRef]
- Fazakas, E.; Zadorozhnyy, V.; Varga, L.K.; Inoue, A.; Louzguine-Luzgin, D.V.; Tian, F.; Vitos, L. Experimental and theoretical study of Ti20Zr20Hf20Nb20X20 (X=V or Cr) refractory high-entropy alloys. Int. J. Refrac. Met. Hard Mater. 2014, 47, 131–138. [Google Scholar] [CrossRef]
- Wang, X.; Xu, S.; Jian, W.R.; Li, X.G.; Su, Y.; Beyerlein, I.J. Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials. Comput. Mater. Sci. 2021, 192, 110364. [Google Scholar] [CrossRef]
- Jian, W.R.; Xu, S.; Beyerlein, I.J. On the significance of model design in atomistic calculations of the Peierls stress in Nb. Comput. Mater. Sci. 2021, 188, 110150. [Google Scholar] [CrossRef]
- Ray, J.R.; Rahman, A. Statistical ensembles and molecular dynamics studies of anisotropic solids. J. Chem. Phys. 1984, 80, 4423–4428. [Google Scholar] [CrossRef]
- Huang, J.; Fang, W.; Xue, C.; Peng, T.; Yu, H.; Li, J.; Sun, L.; He, X.; Liu, B.; Yang, Y.; et al. Exploring the relationship between lattice distortion and phase stability in a multi-principal element alloy system based on machine learning method. Comput. Mater. Sci. 2023, 221, 112089. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, H.; Li, W.; Ding, X.; Wang, Y.; Vitos, L. Generalized Stacking Fault Energy of Al-Doped CrMnFeCoNi High-Entropy Alloy. Nanomaterials 2019, 10, 59. [Google Scholar] [CrossRef]
- Geslin, P.A.; Rodney, D. Microelasticity model of random alloys. Part I: Mean square displacements and stresses. J. Mech. Phys. Solids 2021, 153, 104479. [Google Scholar] [CrossRef]
- Zhang, W.; Chabok, A.; Kooi, B.J.; Pei, Y. Additive manufactured high entropy alloys: A review of the microstructure and properties. Mater. Des. 2022, 220, 110875. [Google Scholar] [CrossRef]
- Feng, R.; Kim, G.; Yu, D.; Chen, Y.; Chen, W.; Liaw, P.K.; An, K. Elastic behavior of binary and ternary refractory multi-principal-element alloys. Mater. Des. 2022, 219, 110820. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, S.; Ding, J.; Chong, Y.; Jia, T.; Ophus, C.; Asta, M.; Ritchie, R.O.; Minor, A.M. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 2020, 581, 283–287. [Google Scholar] [CrossRef]
- Romero, R.A.; Xu, S.; Jian, W.R.; Beyerlein, I.J.; Ramana, C.V. Atomistic simulations of the local slip resistances in four refractory multi-principal element alloys. Int. J. Plast. 2022, 149, 103157. [Google Scholar] [CrossRef]
- Huang, H.; Li, X.; Dong, Z.; Li, W.; Huang, S.; Meng, D.; Lai, X.; Liu, T.; Zhu, S.; Vitos, L. Critical stress for twinning nucleation in CrCoNi-based medium and high entropy alloys. Acta Mater. 2018, 149, 388–396. [Google Scholar] [CrossRef]
- Ding, J.; Yu, Q.; Asta, M.; Ritchie, R.O. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proc. Natl. Acad. Sci. USA 2018, 115, 8919–8924. [Google Scholar] [CrossRef]
- Wang, H.; Pan, R.; Tang, A.; She, J.; Mi, X.; Wu, L.; Tan, J. Effects of Nb concentration and temperature on generalized stacking fault energy of Zr–Nb alloys by molecular dynamics simulations. Mater. Res. Express 2021, 8, 016540. [Google Scholar] [CrossRef]
- Pei, Z.; Zhang, S.; Lei, Y.; Zhang, F.; Chen, M. Decoupling between Shockley partials and stacking faults strengthens multiprincipal element alloys. Proc. Natl. Acad. Sci. USA 2021, 118, e2114167118. [Google Scholar] [CrossRef]
- Zhao, B.; Huang, P.; Zhang, L.; Li, S.; Zhang, Z.; Yu, Q. Temperature Effect on Stacking Fault Energy and Deformation Mechanisms in Titanium and Titanium-aluminium Alloy. Sci. Rep. 2020, 10, 3086. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Xu, S.; Su, Y.; Smith, L.T.W.; Beyerlein, I.J. Frank-Read source operation in six body-centered cubic refractory metals. J. Mech. Phys. Solids 2020, 141, 104017. [Google Scholar] [CrossRef]
- Su, Y.; Ardeljan, M.; Knezevic, M.; Jain, M.; Pathak, S.; Beyerlein, I.J. Elastic constants of pure body-centered cubic Mg in nanolaminates. Comput. Mater. Sci 2020, 174, 109501. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]







| MPEA | Methods | Structures | USFE | ISFE |
|---|---|---|---|---|
| CoCrNi | EAM | Random | 310.2019623 | |
| CSRO | 346.170945 | 82.7860395 | ||
| DFT | Random | 313 [75] | [76] | |
| CSRO | 30 [76] |
| MPEA | Methods | Structures | USFE |
|---|---|---|---|
| MoNbTa | EAM | Random | 865.43 |
| EAM | CSRO | 858.12 | |
| DFT | Random | 1054.58106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubassira, S.; Jian, W.-R.; Xu, S. Effects of Chemical Short-Range Order and Temperature on Basic Structure Parameters and Stacking Fault Energies in Multi-Principal Element Alloys. Modelling 2024, 5, 352-366. https://doi.org/10.3390/modelling5010019
Mubassira S, Jian W-R, Xu S. Effects of Chemical Short-Range Order and Temperature on Basic Structure Parameters and Stacking Fault Energies in Multi-Principal Element Alloys. Modelling. 2024; 5(1):352-366. https://doi.org/10.3390/modelling5010019
Chicago/Turabian StyleMubassira, Subah, Wu-Rong Jian, and Shuozhi Xu. 2024. "Effects of Chemical Short-Range Order and Temperature on Basic Structure Parameters and Stacking Fault Energies in Multi-Principal Element Alloys" Modelling 5, no. 1: 352-366. https://doi.org/10.3390/modelling5010019
APA StyleMubassira, S., Jian, W. -R., & Xu, S. (2024). Effects of Chemical Short-Range Order and Temperature on Basic Structure Parameters and Stacking Fault Energies in Multi-Principal Element Alloys. Modelling, 5(1), 352-366. https://doi.org/10.3390/modelling5010019