
A Molecular Electron Density Theory Study of the [3+2] Cycloaddition Reaction of Pseudo(mono)radical Azomethine Ylides with Phenyl Vinyl Sulphone




Abstract
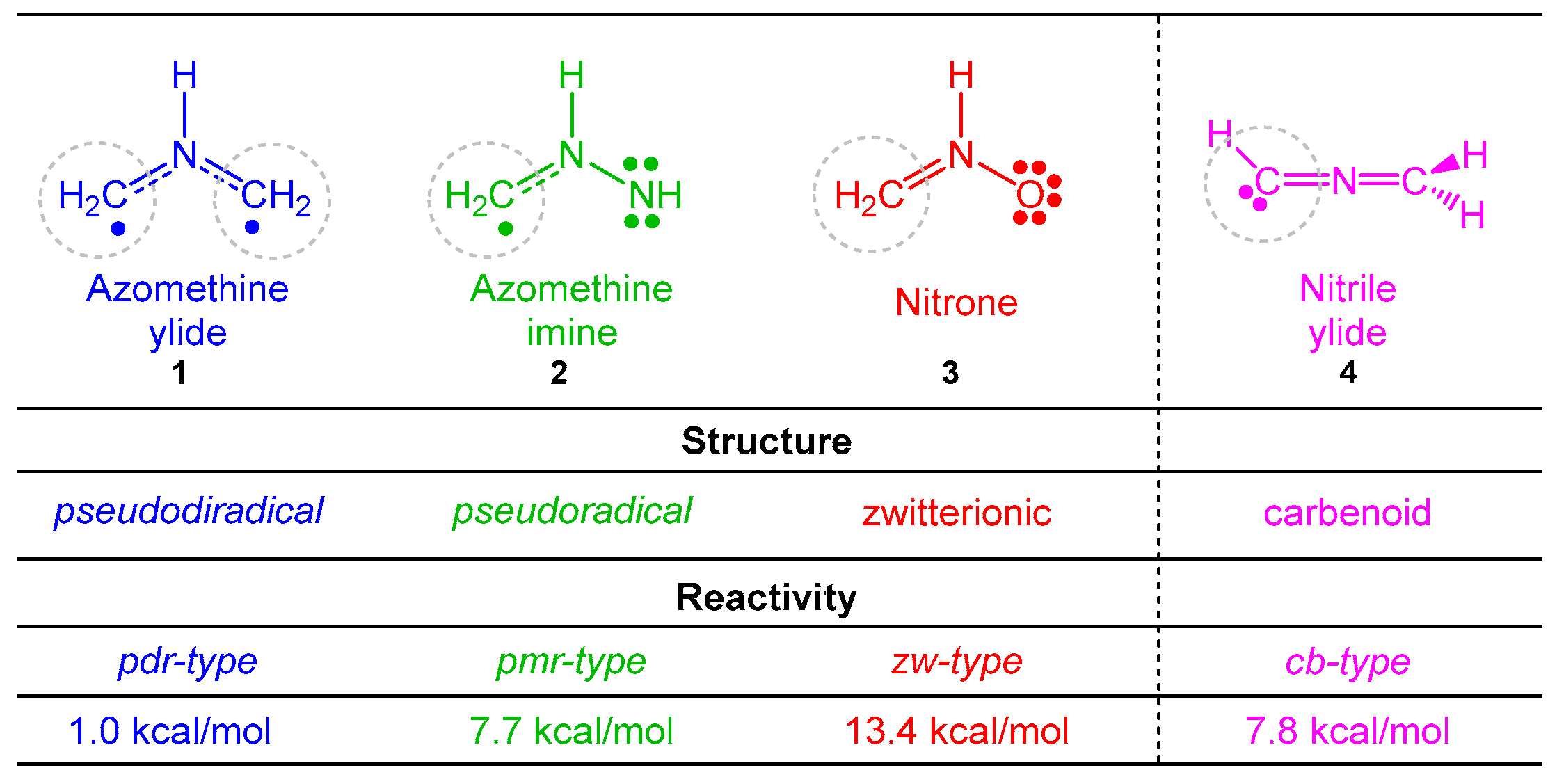
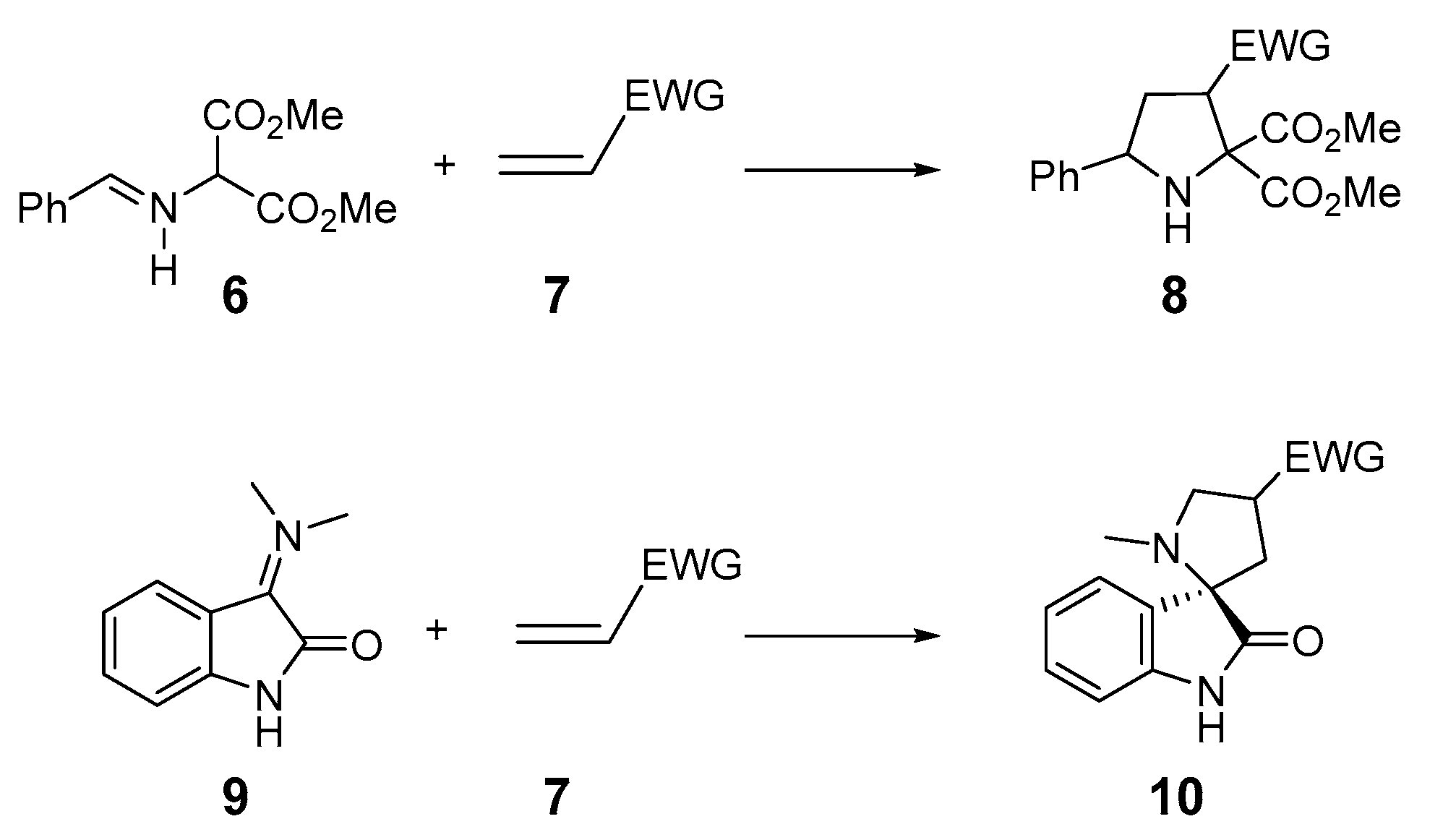
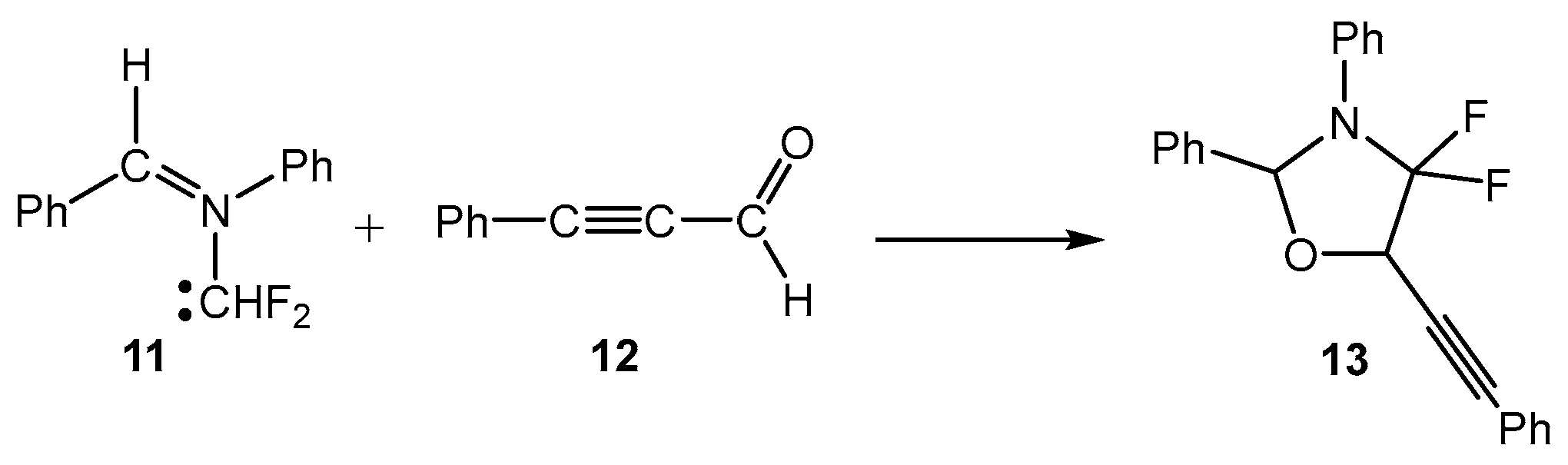
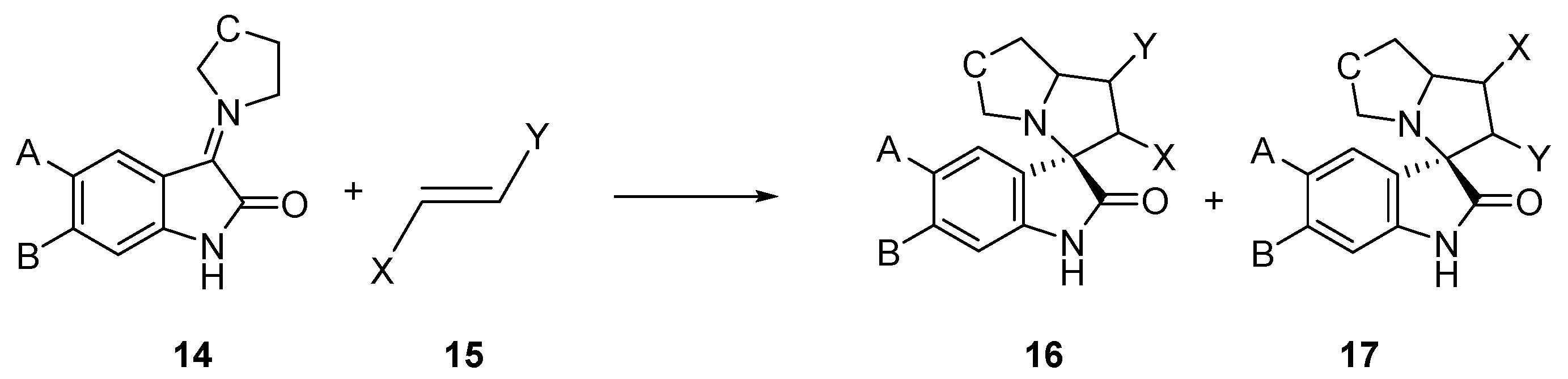
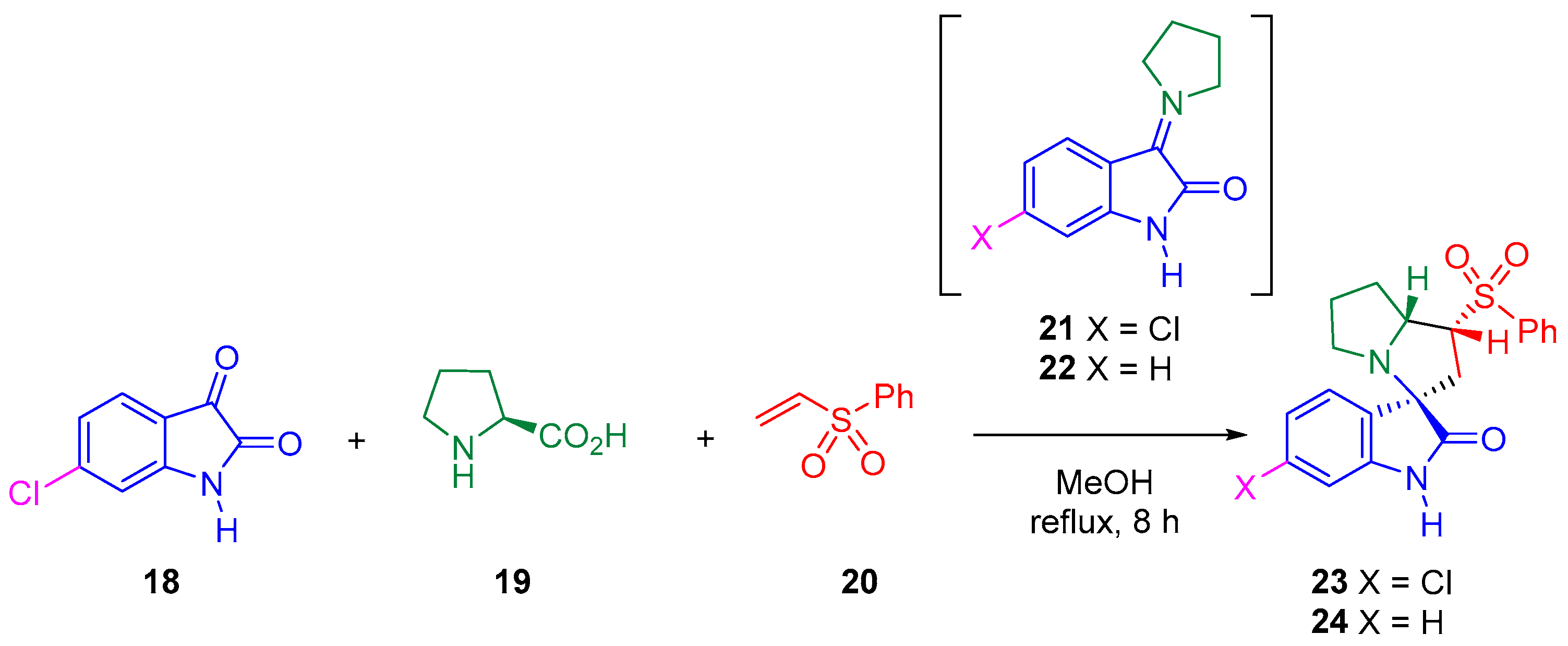
:1. Introduction
2. Materials and Methods
3. Results and Discussion
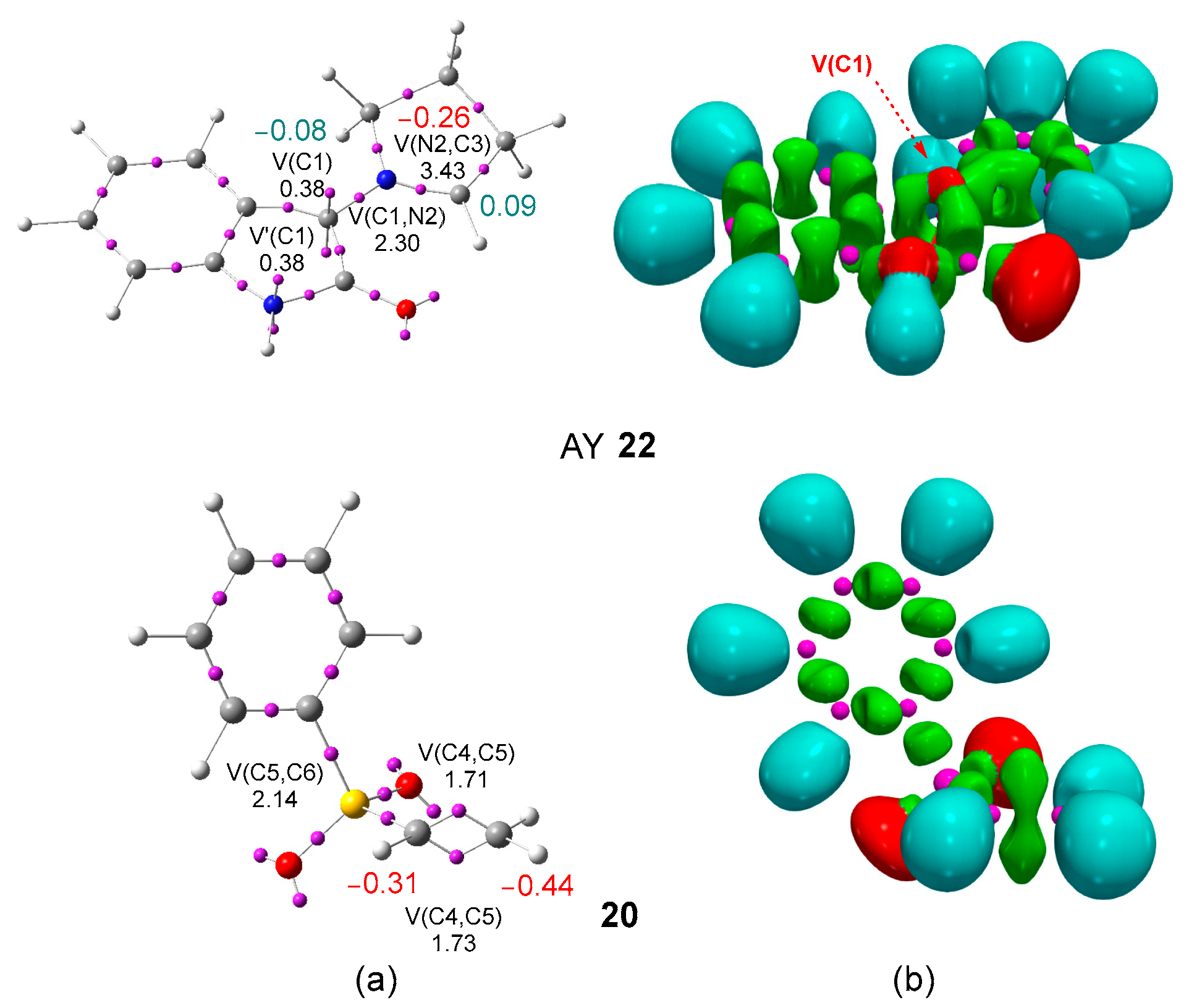
3.1. ELF Topological Analysis at the Ground State of AY 22 and Phenyl Vinyl Sulphone 20
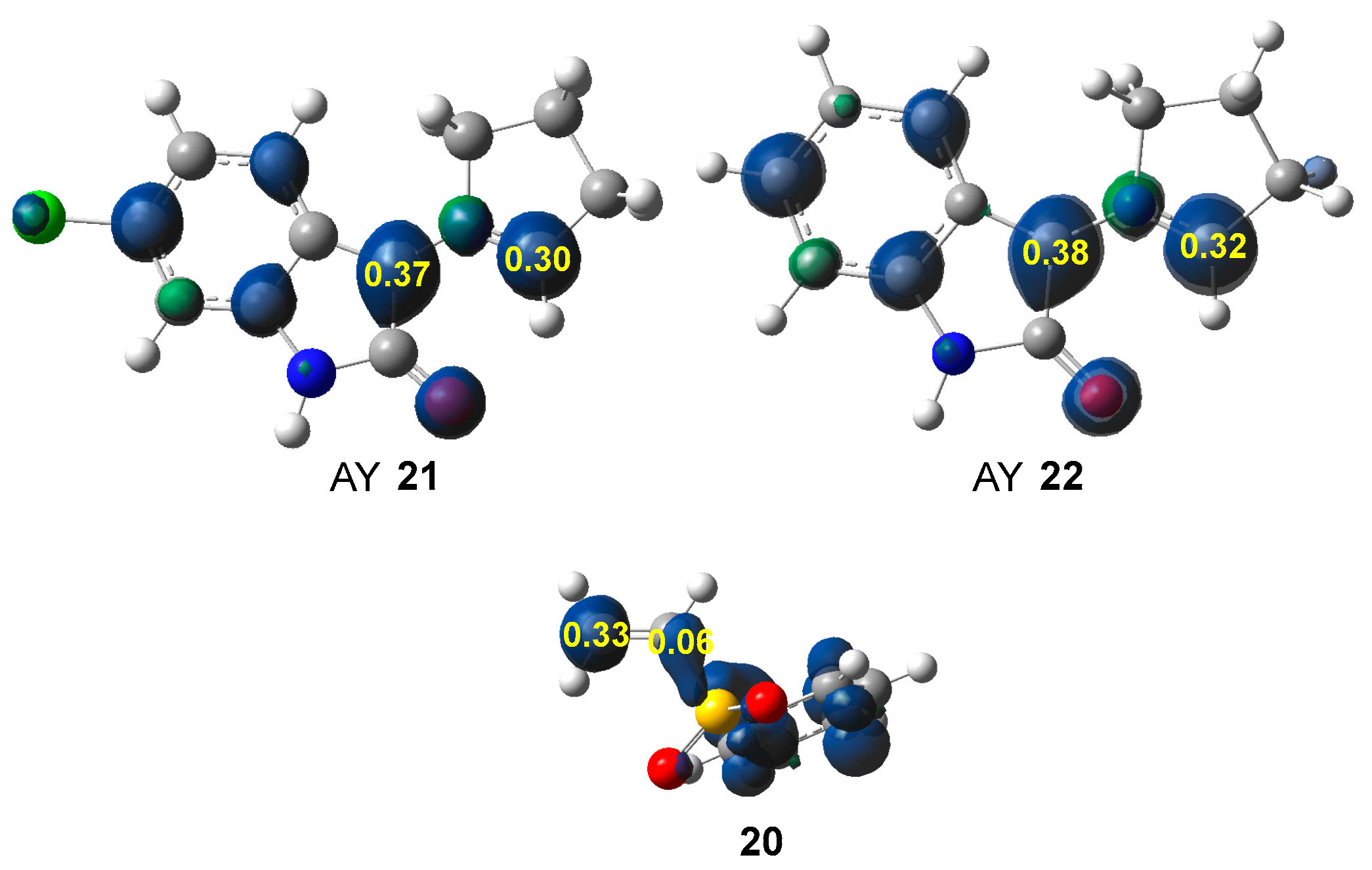
3.2. Conceptual DFT Analysis at the Ground State of the Reagents
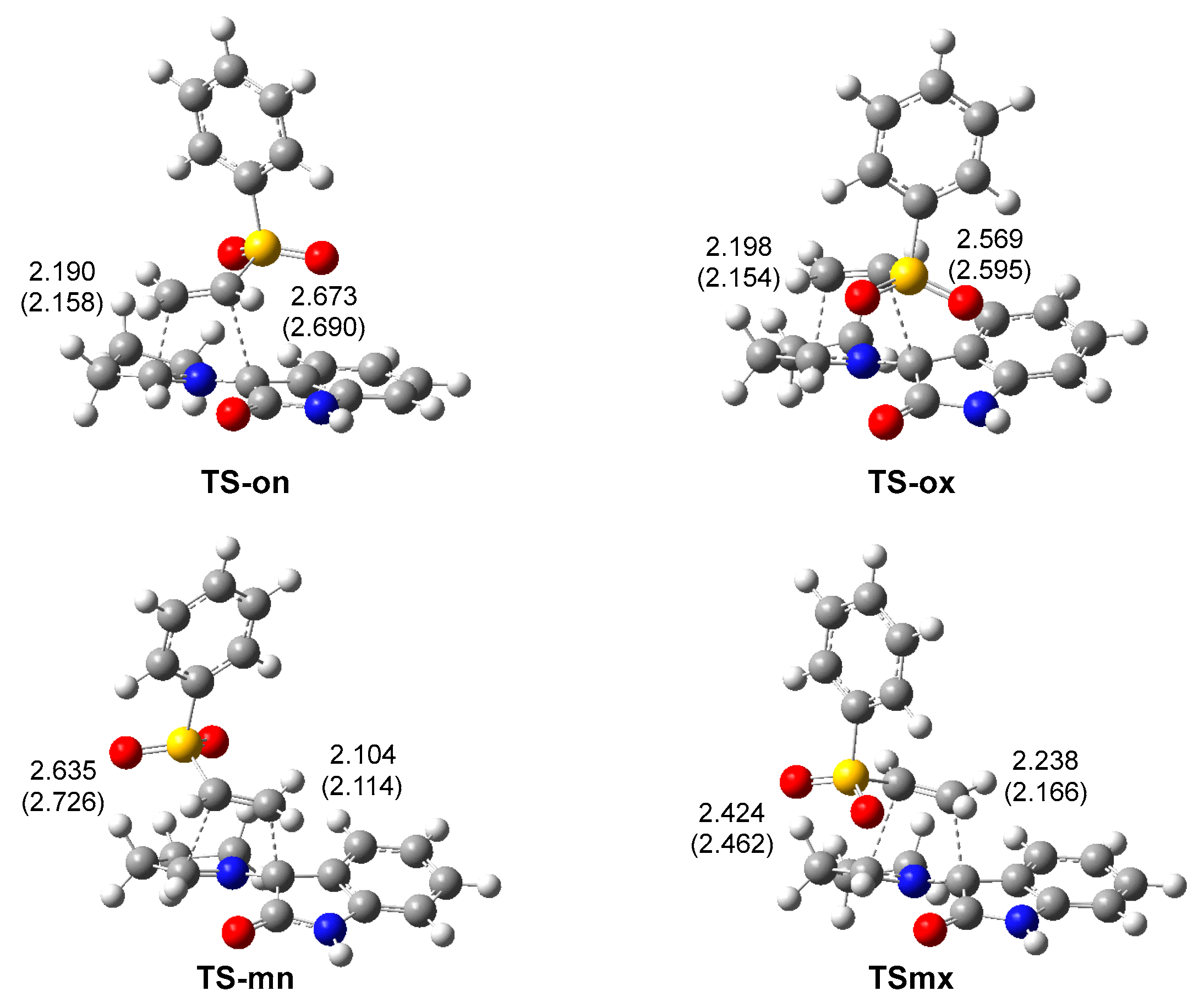
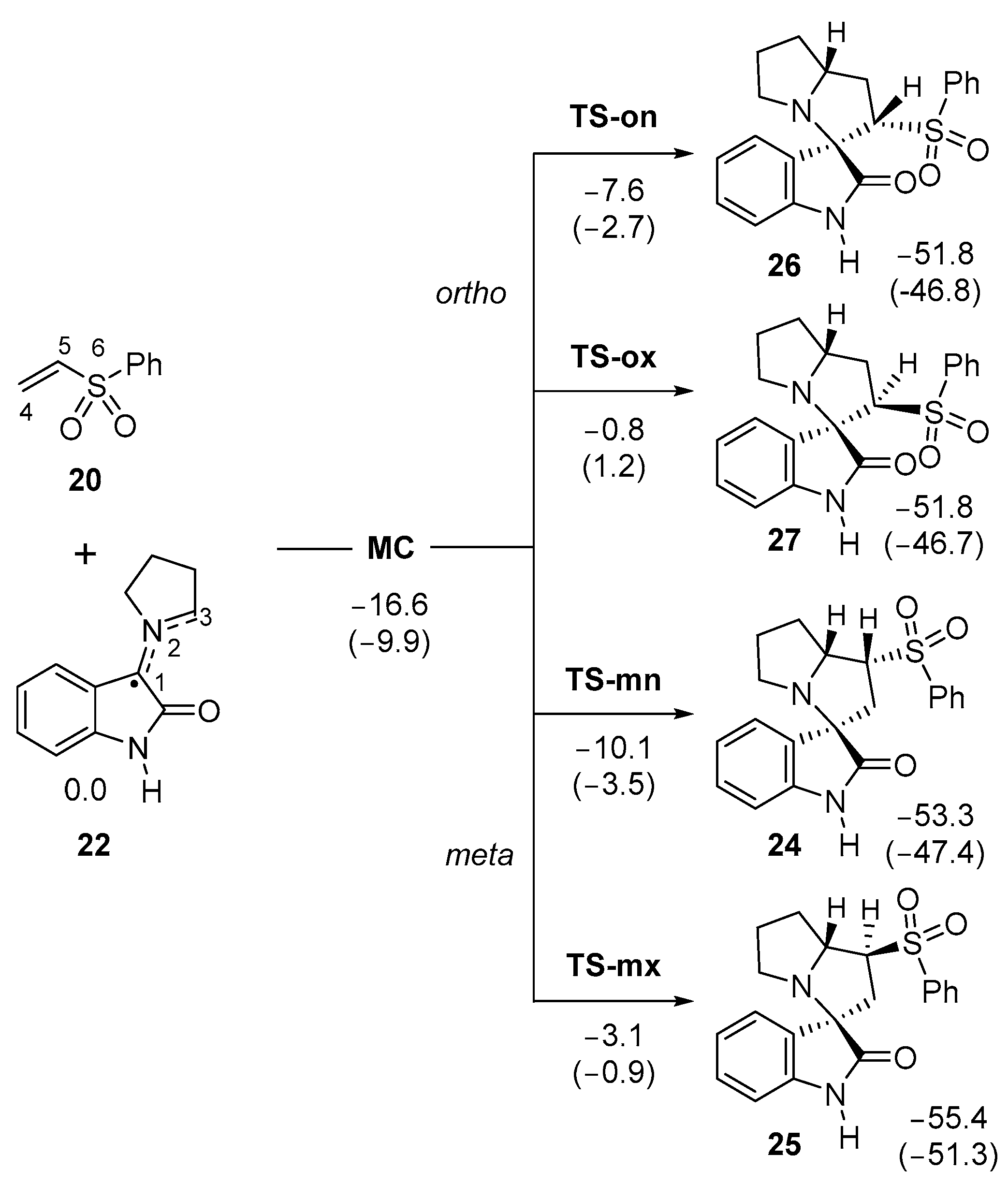
3.3. Study of the Reaction Paths Associated with the 32CA Reaction of AY 22 with Phenyl Vinyl Sulphone 20
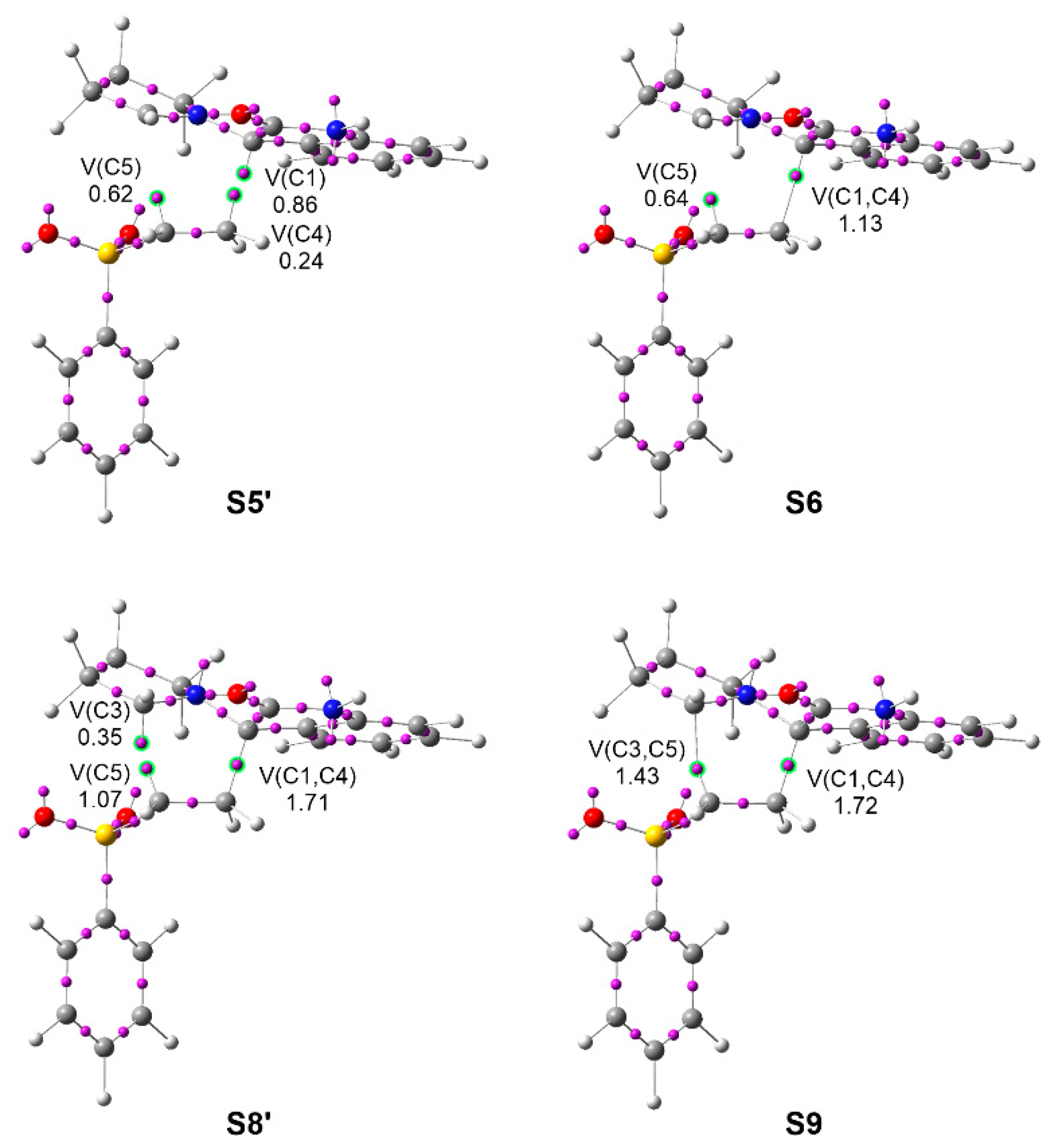
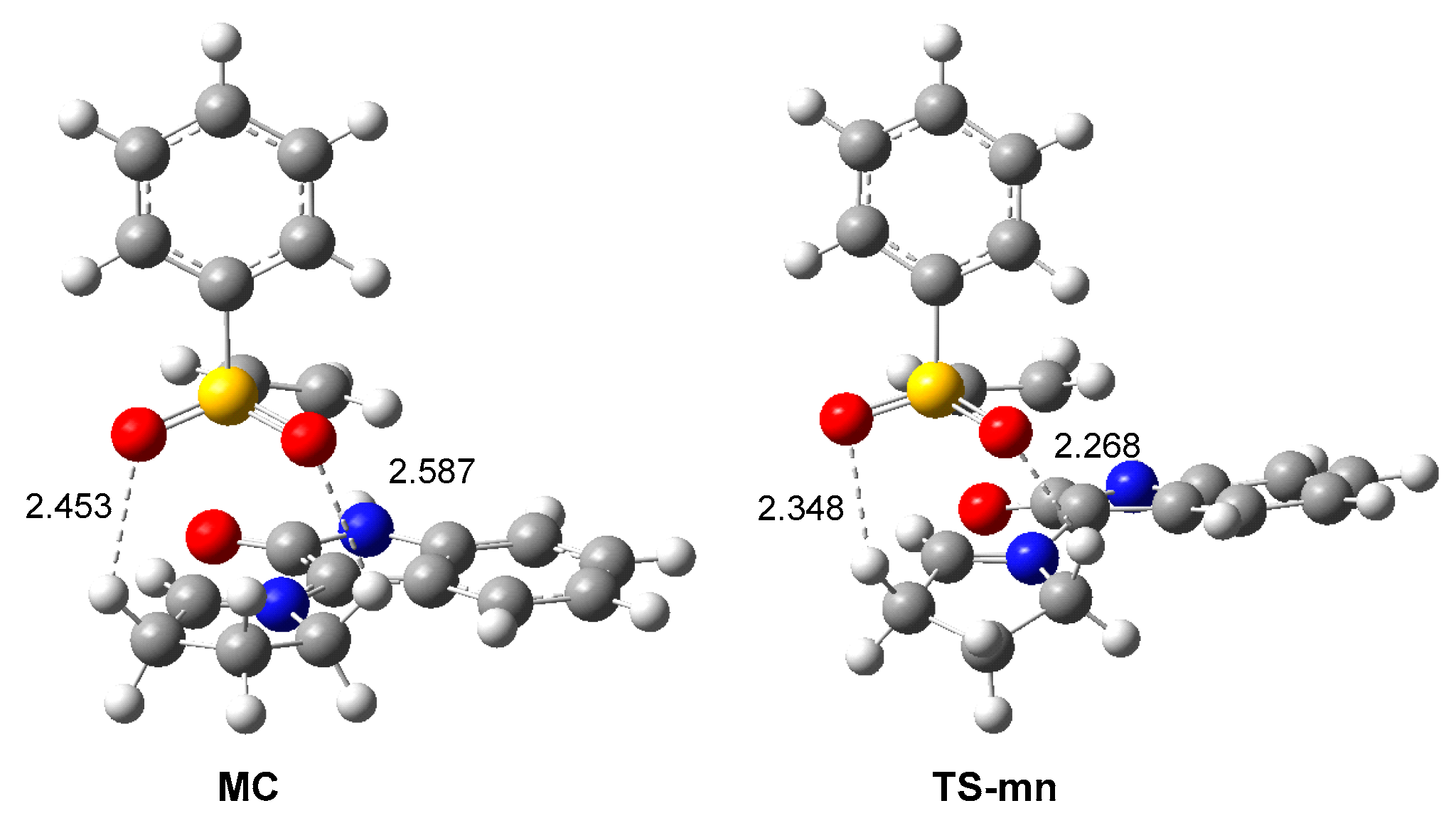
3.4. BET Analysis along the Most Favorable Meta/Endo Reaction Path. Characterization of the Pmr-Type Mechanism
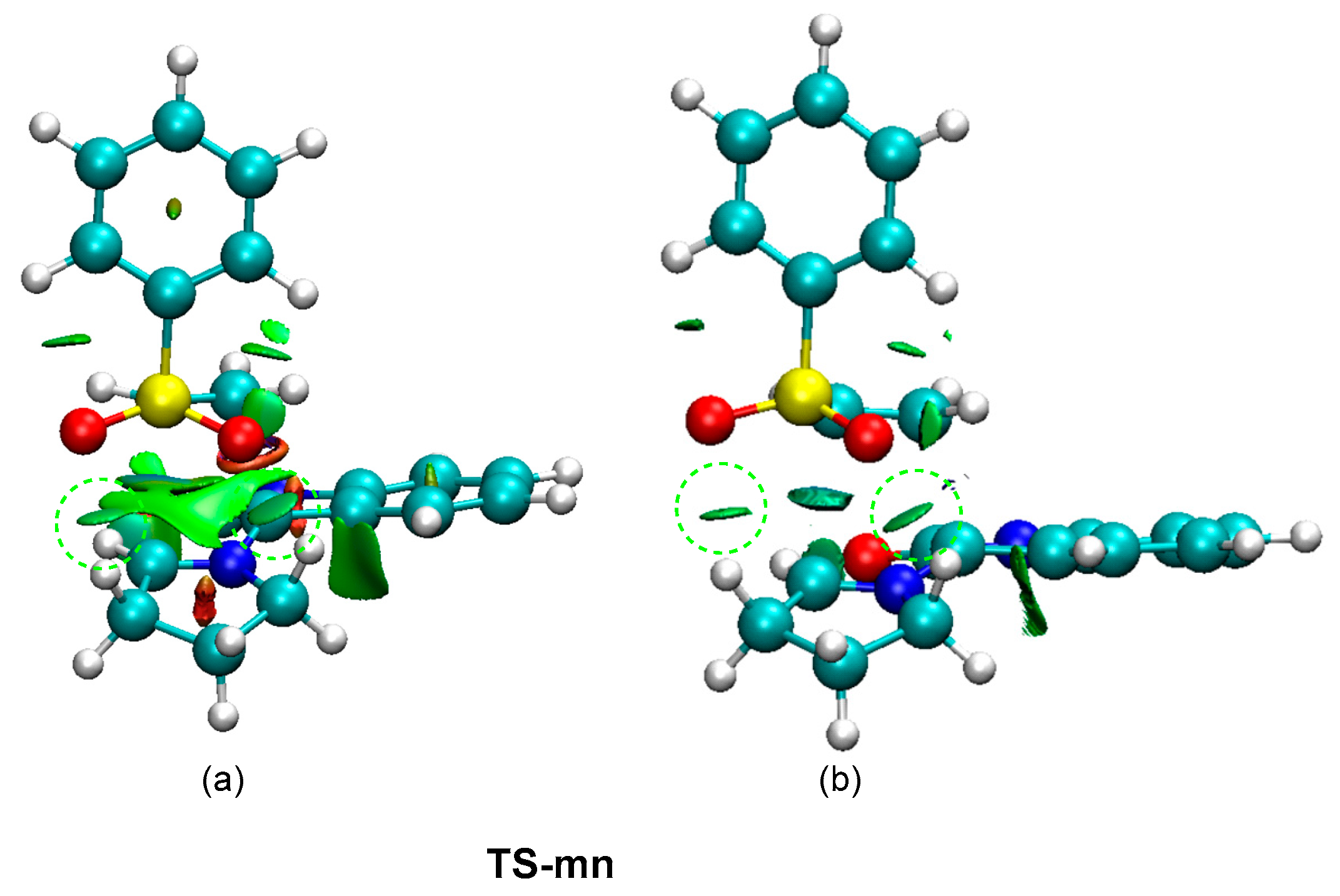
3.5. Analysis of the Origin of the Meta Regio- and Endo Stereo-Selectivities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carruthers, W. Some Modern Methods of Organic Synthesis, 2nd ed.; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Padwa, A. 1,3-Dipolar Cycloaddition Chemistry; Wiley-Interscience: New York, NY, USA, 1984; Volumes 1–2. [Google Scholar]
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1990. [Google Scholar]
- Padwa, A.; Pearson, W.H. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons, Inc.: New York, NY, USA, 2002; Volume 59. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the High Reactivity of the Azomethine Ylides in [3+2] Cycloaddition Reactions. Lett. Org. Chem. 2010, 7, 432–439. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions. Molecules 2017, 22, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Reactivity and Selectivities in [3+2] Cycloaddition Reactions of C,N-Dialkyl Nitrones with Ethylene Derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. An MEDT study of the carbenoid-type [3+2] cycloaddition reactions of nitrile ylides with electron-deficient chiral oxazolidinones. Org. Biomol. Chem. 2016, 14, 10427–10436. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 2019, 267–282. [Google Scholar] [CrossRef]
- Bailly, C. Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 363–378. [Google Scholar] [CrossRef]
- Bellina, F.; Rossi, R. Synthesis and biological activity of pyrrole, pyrroline and pyrrolidine derivatives with two aryl groups on adjacent positions. Tetrahedron 2006, 62, 7213–7256. [Google Scholar] [CrossRef]
- Narayan, R.; Potowski, M.; Jia, Z.-J.; Antonchick, A.P.; Waldmann, H. Catalytic Enantioselective 1,3-Dipolar Cycloadditions of Azomethine Ylides for Biology-Oriented Synthesis. Acc. Chem. Res. 2014, 47, 1296–1310. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Ali, M.; Al-Majid, A.M.; Alshahrani, S.; Alamary, A.S.; Yousuf, S.; Choudhary, M.I. Regio- and Stereoselective Synthesis of a New Series of Spirooxindole Pyrrolidine Grafted Thiochromene Scaffolds as Potential Anticancer Agents. Symmetry 2021, 13, 1426. [Google Scholar] [CrossRef]
- Boudriga, S.; Haddad, S.; Murugaiyah, V.; Askri, M.; Knorr, M.; Strohmann, C.; Golz, C. Three-Component Access to Functionalized Spiropyrrolidine Heterocyclic Scaffolds and Their Cholinesterase Inhibitory Activity. Molecules 2020, 25, 1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, A.; Soliman, S.M.; Alshahrani, S.; Islam, M.S.; Ali, M.; Al-Majid, A.M.; Yousuf, S. Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo [b] Thiophene Analogue. Crystals 2020, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; Alshahrani, S.; Al-Majid, A.M.; Ali, M.; Altowyan, M.S.; Islam, M.S.; Alamary, A.S.; Ashraf, S.; Ul-Haq, Z. Synthesis of a New Class of Spirooxindole–Benzo [b] Thiophene-Based Molecules as Acetylcholinesterase Inhibitors. Molecules 2020, 25, 4671. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Lotfy, G.; Ali, M.; Mostafa, A.; Elshaier, Y.A.; Al-Habib, S. Drug Repurposing of Lactoferrin Combination in a Nanodrug Delivery System to Combat Severe Acute Respiratory Syndrome Coronavirus-2 Infection. Med. J. 2021, 3, 104–112. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Electronic Reorganization along the Nonpolar [3+2] Cycloaddition Reactions of Carbonyl Ylides. J. Org. Chem. 2011, 76, 373–379. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Role of the Copper Metalation of Azomethine Ylides in [3+2] Cycloaddition Reactions. J. Org. Chem. 2018, 83, 10959–10973. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. A mechanistic study of the participation of azomethine ylides and carbonyl ylides in [3+2] cycloaddition reactions. Tetrahedron 2015, 71, 1050–1057. [Google Scholar] [CrossRef]
- Aziz, Y.M.A.; Lotfy, G.; Said, M.M.; El Ashry, E.S.H.; El Tamany, E.S.H.; Soliman, S.M.; Abu-Serie, M.M.; Teleb, M.; Yousuf, S.; Dömling, A.; et al. Design, Synthesis, Chemical and Biochemical Insights Into Novel Hybrid Spirooxindole-Based p53-MDM2 Inhibitors With Potential Bcl2 Signaling Attenuation. Front. Chem. 2021, 9, 735236. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; Azam, M.; Verma, V.P.; Barakat, A.; Haukka, M.; Domingo, L.R.; Elgazar, A.A.; Mira, A.; Badria, F.A. Synthesis of Spirooxindole Analogs Tethered Pyrazole Scaffold as Acetylcholinesterase Inhibitors. Chem. Sel. 2021, 6, 14039–14053. [Google Scholar]
- Barakat, A.; Haukka, M.; Soliman, S.M.; Ali, M.; Al-Majid, A.M.; El-Faham, A.; Domingo, L.R. Straightforward Regio- and Diastereoselective Synthesis, Molecular Structure, Intermolecular Interactions and Mechanistic Study of Spirooxindole-Engrafted Rhodanine Analogs. Molecules 2021, 26, 7276. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Soliman, S.M.; Haukka, M.; Ali, M.; Islam, M.S.; Shaik, M.R.; Barakat, A. Design, Construction, and Characterization of a New Regioisomer and Diastereomer Material Based on the Spirooxindole Scaffold Incorporating a Sulphone Function. Symmetry 2020, 12, 1337. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehre, M.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions–Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, 6th ed.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theo. Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Reactivity of Tetrazines in Aza- Diels-Alder Reactions. RSC Adv. 2020, 10, 15394–15405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, E.; Duque-Noreña, M.; Gutiérrez-Sánchez, N.; Rincón, E.; Domingo, L.R. A Close Look to the Oxaphosphetane Formation along the Wittig Reaction: A [2 + 2] Cycloaddition? J. Org. Chem. 2020, 85, 6675–6686. [Google Scholar] [CrossRef]
- Aurell, M.J.; Domingo, L.R.; Perez, P.; Contreras, R. A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Sáez, J.A. Understanding the Origin of the Asynchronicity in Bond-Formation in Polar Cycloaddition Reactions. A DFT Study of the 1,3-Dipolar Cycloaddition Reaction of Carbonyl Ylides with 1,2-Benzoquinones. RSC Adv. 2012, 2, 1334–1342. [Google Scholar] [CrossRef]
- Benchouk, W.; Mekelleche, S.M.; Silvi, B.; Aurell, M.J.; Domingo, L.R. Understanding the kinetic solvent effects on the 1,3-dipolar cycloaddition of benzonitrile N-oxide: A DFT study. J. Phys. Org. Chem. 2011, 24, 611–618. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar Cycloadditions toward Electrophilic Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. Molecular Orbitals in Chemistry, Physics, and Biology; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Gleiter, R.; Bohm, M.C. Regio- and Stereoselectivity in Diels-Alder Reactions. Theoretical Considerations. Pure Appl. Chem. 1983, 55, 237–244. [Google Scholar] [CrossRef]
- Ramírez, M.; Svatunek, D.; Liu, F.; Garg, N.K.; Houk, K.N. Origins of Endo Selectivity in Diels–Alder Reactions of Cyclic Allene Dienophiles. Angew. Chem. Int. Ed. 2021, 60, 14989–14997. [Google Scholar] [CrossRef]
- García, J.I.; Mayoral, J.A.; Salvatella, L. Do Secondary Orbital Interactions Really Exist? Acc. Chem. Res. 2000, 33, 658–664. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| vinyl sulphone 20 | −4.39 | 5.97 | 1.61 | 1.75 |
| AY 21 | −2.97 | 3.34 | 1.32 | 4.48 |
| AY 22 | −2.76 | 3.36 | 1.14 | 4.68 |
| ∆H | ∆S | ∆G | |
|---|---|---|---|
| MC | −7.6 | −32.8 | 3.4 |
| TS-on | −1.1 | −46.7 | 14.7 |
| TS-ox | 2.8 | −44.0 | 17.6 |
| TS-mn | −1.9 | −44.4 | 13.1 |
| TS-mx | 0.7 | −43.0 | 15.2 |
| 26 | −42.4 | −51.2 | −25.1 |
| 27 | −42.7 | −50.6 | −25.6 |
| 24 | −43.3 | −48.7 | −26.8 |
| 25 | −47.2 | −48.5 | −30.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos-Gutiérrez, M.; Barakat, A.; Domingo, L.R. A Molecular Electron Density Theory Study of the [3+2] Cycloaddition Reaction of Pseudo(mono)radical Azomethine Ylides with Phenyl Vinyl Sulphone. Organics 2022, 3, 122-136. https://doi.org/10.3390/org3020010
Ríos-Gutiérrez M, Barakat A, Domingo LR. A Molecular Electron Density Theory Study of the [3+2] Cycloaddition Reaction of Pseudo(mono)radical Azomethine Ylides with Phenyl Vinyl Sulphone. Organics. 2022; 3(2):122-136. https://doi.org/10.3390/org3020010
Chicago/Turabian StyleRíos-Gutiérrez, Mar, Assem Barakat, and Luis R. Domingo. 2022. "A Molecular Electron Density Theory Study of the [3+2] Cycloaddition Reaction of Pseudo(mono)radical Azomethine Ylides with Phenyl Vinyl Sulphone" Organics 3, no. 2: 122-136. https://doi.org/10.3390/org3020010
APA StyleRíos-Gutiérrez, M., Barakat, A., & Domingo, L. R. (2022). A Molecular Electron Density Theory Study of the [3+2] Cycloaddition Reaction of Pseudo(mono)radical Azomethine Ylides with Phenyl Vinyl Sulphone. Organics, 3(2), 122-136. https://doi.org/10.3390/org3020010