1. Introduction
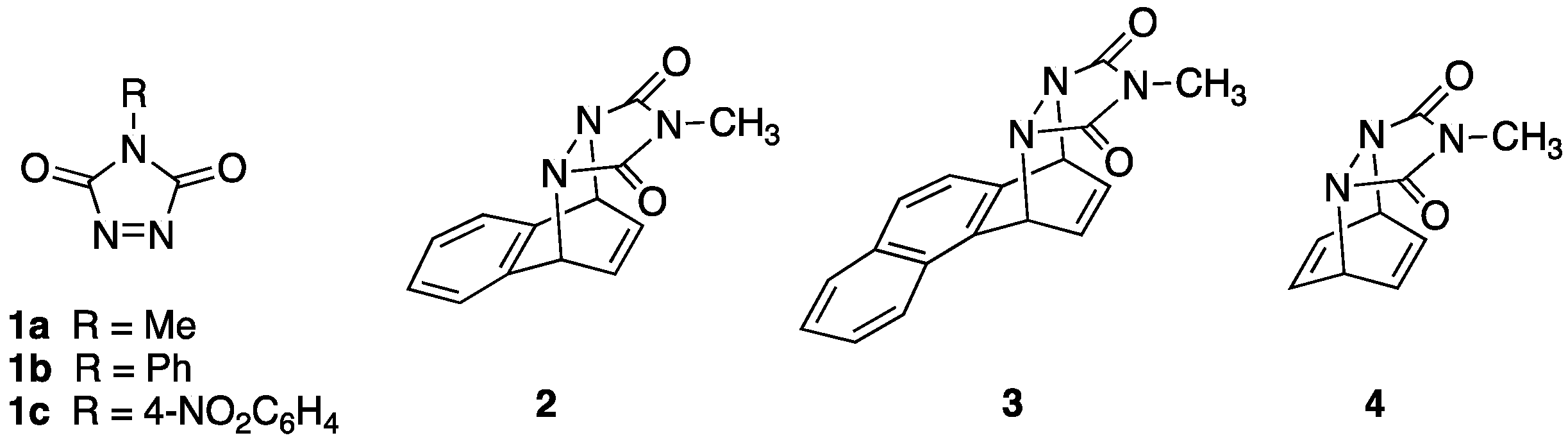
N-Substituted 1,2,4-triazoline-3,5-diones (TADs,
1, see
Figure 1) are highly electrophilic azo compounds that undergo thermal reactions with many classes of organic compounds, including alcohols, alkenes, alkynes, and aromatic substrates [
1]. TADs are generally deep red in color, rendering them susceptible to further activation via visible-light irradiation [
2]. Indeed, under photochemical conditions, the reactivity of TADs is known to be even further enhanced, allowing for many reactions that will not occur thermally, as for example, reaction with strained saturated C-C bonds and aromatic substrates [
2,
3,
4].
In a series of seminal papers, Sheridan demonstrated that
N-methyl-1,2,4-triazoline-3,5-dione (MeTAD,
1a) undergoes photochemical Diels–Alder (DA) type cycloaddition reactions with naphthalene, phenanthrene, and even benzene, to form cycloadducts
2,
3, and
4, respectively [
5,
6,
7,
8]. While further studies in our lab demonstrated that naphthalene exhibited some thermal reactivity with
1a to form
2 [
9], photochemical conditions drove the reaction to completion faster, and provided a higher isolated yield. On the contrary, however, no reaction between
1a and either phenanthrene or benzene is observed in the absence of light [
7,
8]. In addition, whereas reaction with naphthalene and phenanthrene occurs at room temperature, the photochemical reaction with benzene was reported to take place only at temperatures below –60 °C, and no reaction at room temperature [
5,
8].
The photochemically driven Diels–Alder reaction of
1a with benzene is of particular significance given the general reluctance of benzene to undergo any sort of addition reaction because of the resulting loss of aromatic stabilization, although exceptions have been noted [
10]. Wamhoff first recorded the ability to engage benzene in photoreactivity with a triazolinedione (using the especially electrophilic
N-4-nitrophenyl derivative,
1c) in 1977 [
2]. Sheridan’s work more clearly documented the course of such reactions in 1989 [
8]. More recently, Sarlah’s group (2016–current) has exploited the photo-driven Diels–Alder reactivity of aromatics with MeTAD for fascinating synthetic applications [
11,
12].
Given the recent resurgence in both the interest and applications of this reaction, in this report, a related finding is described in which a novel adduct resulting from a double electrophilic aromatic substitution reaction was observed during the photochemical reaction of 1a with benzene at room temperature.
2. Results
During the course of a previous study on the room temperature photochemical reactions of
1a with variously substituted benzenes [
4], a reinvestigation of the reaction of
1a with benzene was undertaken. The results of these reactions are summarized in
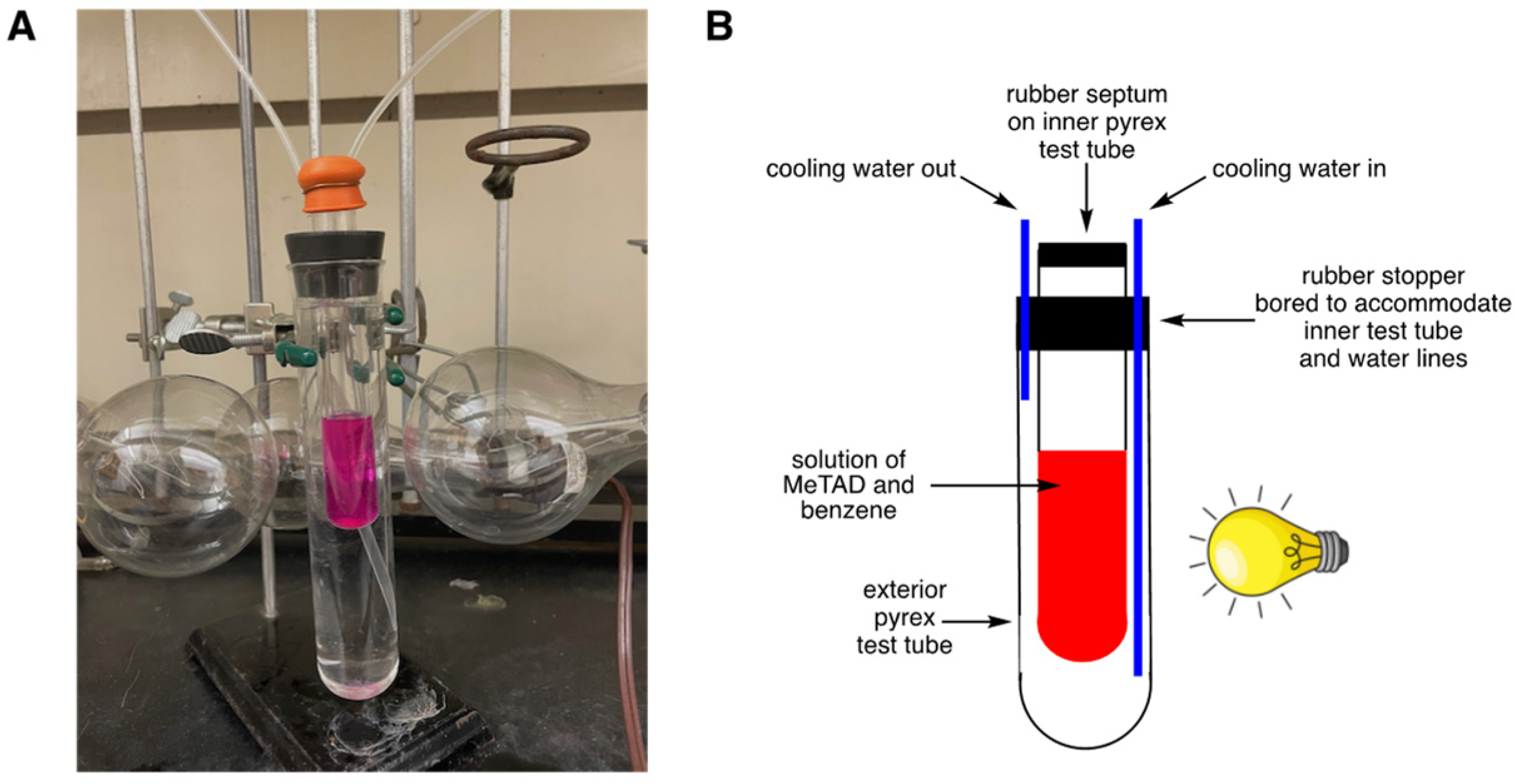
Table 1. Thus, within 3 h of visible light (3 × 300 W incandescent bulbs) irradiation of 10 mL of a red-colored solution of
1a and benzene (0.1 M each) in CH
2Cl
2, some crystals were observed to form on the sides of the reaction vessel. After irradiating for a total of 24 h, the crystals needed to be scraped from the sides and allowed to settle to the bottom of the reaction flask, in order to admit sufficient light to continue the photochemical reaction. After 48 h of irradiation, additional crystals formed, and the solution turned to a very pale pink color, indicating the near complete consumption of
1a. The crystals were isolated via vacuum filtration. Analysis of the crystals by
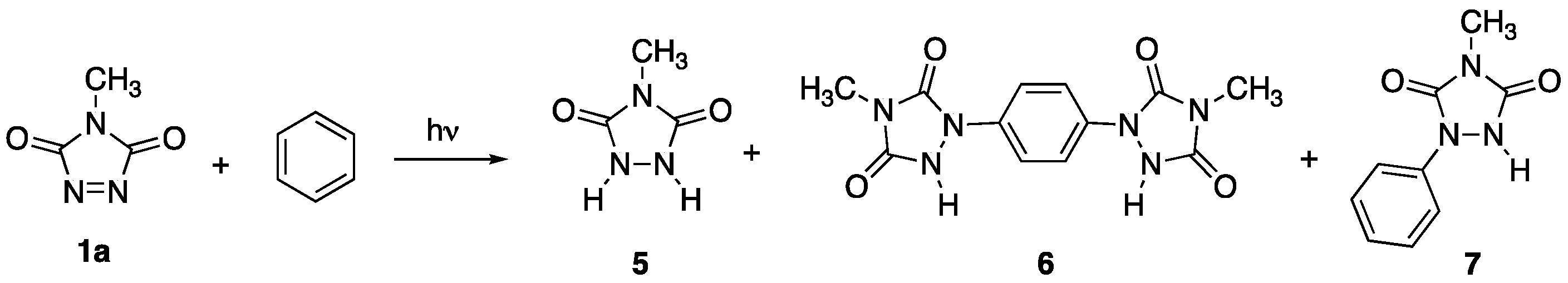
1H NMR spectroscopy indicated the presence of a ~2:1 mixture of
N-methylurazole
5 (
Scheme 1) and an additional novel compound. This novel compound was insoluble in methanol (whereas
5 has appreciable solubility) so separation of the two compounds proved to be operatively simple. It was calculated that 30% of
1a had been converted to
5 (
Table 1, entry 1).
Analysis of the novel compound by
1H NMR,
13C NMR, IR, and HRMS revealed the structure to be that of the
para-substituted bisurazole compound
6 (see spectra provided in the
Supplementary Materials). Only a single
N-methyl signal was observed in the
1H NMR spectrum, but that would be consistent with the
ortho- and
meta-substituted products as well, due to the symmetry of the various possible regioisomers. However, only a single singlet was observed for all four of the aromatic protons in the
1H NMR spectrum, and only two carbon signals for the six benzene ring carbons in the
13C NMR spectrum. These data are only consistent with the
para-substituted structure of
6. This compound was formed in 17% yield (
Table 1, entry 1).
Concentration of the filtrate from above, and examination of its contents by
1H NMR spectroscopy revealed the presence of photodegradation products of
1a, and trace amounts of the known monosubstituted urazole
7. Such photodegradation of TADs in solution has been documented before [
2,
13]. It should be noted that Wamhoff had reported the formation of a monosubstituted urazole comparable to
7 upon irradiation of
1c with benzene [
2]. He did not, however, observe the formation of a bis-adduct.
In an attempt to increase the rate of reaction, the amount of benzene in the reaction was increased to five equivalents relative to
1a, but under otherwise identical conditions (see
Table 1, entry 2). Interestingly, the reaction proceeded even slower in the presence of excess benzene, and the solution remained deep purple-red in color even after 2 days of irradiation. Furthermore, the combined yield of
5 and
6 isolated as crystals (26 mg) was less than that when equimolar amounts of the two starting materials had been irradiated (60 mg). Concentration of the remaining filtrate afforded unreacted MeTAD, small amounts of
7, and photodegradation products.
It was suspected that the
N-methylurazole
5 that was formed might be a result of the reaction of photoactive
1a with CH
2Cl
2 with the solvent acting as an H-donor. Therefore, the solvent was changed to CH
3CN and the reaction was then carried out under otherwise identical conditions (
Table 1, entry 3). The reaction took 3 days to complete, as opposed to the 2 days in CH
2Cl
2, but the only product observed was bisurazole
6 and in a higher isolated yield of 53%. Concentration of the filtrate revealed the presence of small amounts (<10%) of monosubstituted
7 in addition to photodegradation products. Next, a control reaction was carried out for a period of only 24 h such that the reaction was intentionally not allowed to run to completion (
Table 1, entry 4). Under these conditions, bis-adduct
6 was isolated in 16% yield and monosubstituted
7 was obtained in 10% yield.
As a control experiment to definitively determine that bisurazole 6 was formed from further reaction of monosubstituted 7, MeTAD was irradiated in 10 mL of a saturated acetonitrile solution of 7, but otherwise in the absence of any added benzene. As before, crystal formation was observed to take place within a few hours of irradiation. After 24 h, bis-adduct 6 was isolated in 65% yield, confirming that final product 6 is formed via initially generated 7.
Finally, it is interesting to note that an attempt to carry out this reaction with the N-phenyl TAD derivative PhTAD (1b) in CH3CN did not lead to the formation of either mono or disubstituted adducts even after carrying out the irradiation for a total of 4 days. Indeed, at least 80% of 1b remained present in solution at the end of the reaction period.
3. Discussion
During initial studies of the photochemical reaction of MeTAD with various aromatic compounds (presumably at room temperature), Sheridan reported that his group was unable to observe any reaction with benzene or several substituted benzenes [
5]. However, he reported that successful Diels–Alder reactivity with benzene to form
4 was readily observed when the photochemical reaction was conducted at temperatures below −60 °C (in benzene as solvent) [
8]. Raising the temperature to even −10 °C was sufficient to trigger a retro-Diels–Alder reaction to occur, converting the cycloadduct back to starting materials (t
1/2 = 1 h at 0 °C). The lack of observed reactivity at room temperature, therefore, could be explained by the thermal instability of the cycloadduct. In other words, although photochemical cycloaddition actually occurred at room temperature to form
4, the cycloaddition product quickly reverted to starting materials via the thermally driven retro-DA process, thereby providing an illusion of no reaction. Therefore, it was fortunate that under the experimental conditions of this study, the beginnings of crystal formation on the sides of the reaction vessel had been observed, because formation of these crystals prompted the continued irradiation of the solution for an extended period of time.
It is interesting that even when the photochemical reaction conducted in CH
3CN was interrupted prior to the complete consumption of
1a (
Table 1, entry 4), the amount of disubstituted product
6 was in excess of the monosubstituted
7. The preference for the formation of
6 was also the case for the reaction conducted in CH
2Cl
2 in the presence of an excess of benzene. Such conditions might be expected to favor the formation of the monosubstituted adduct over the disubstituted adduct. Therefore, monosubstituted
7 must be more reactive toward photoactivated
1a than benzene itself. This behavior is consistent with the urazole ring acting as an activating (electron-donating) substituent on the ring.
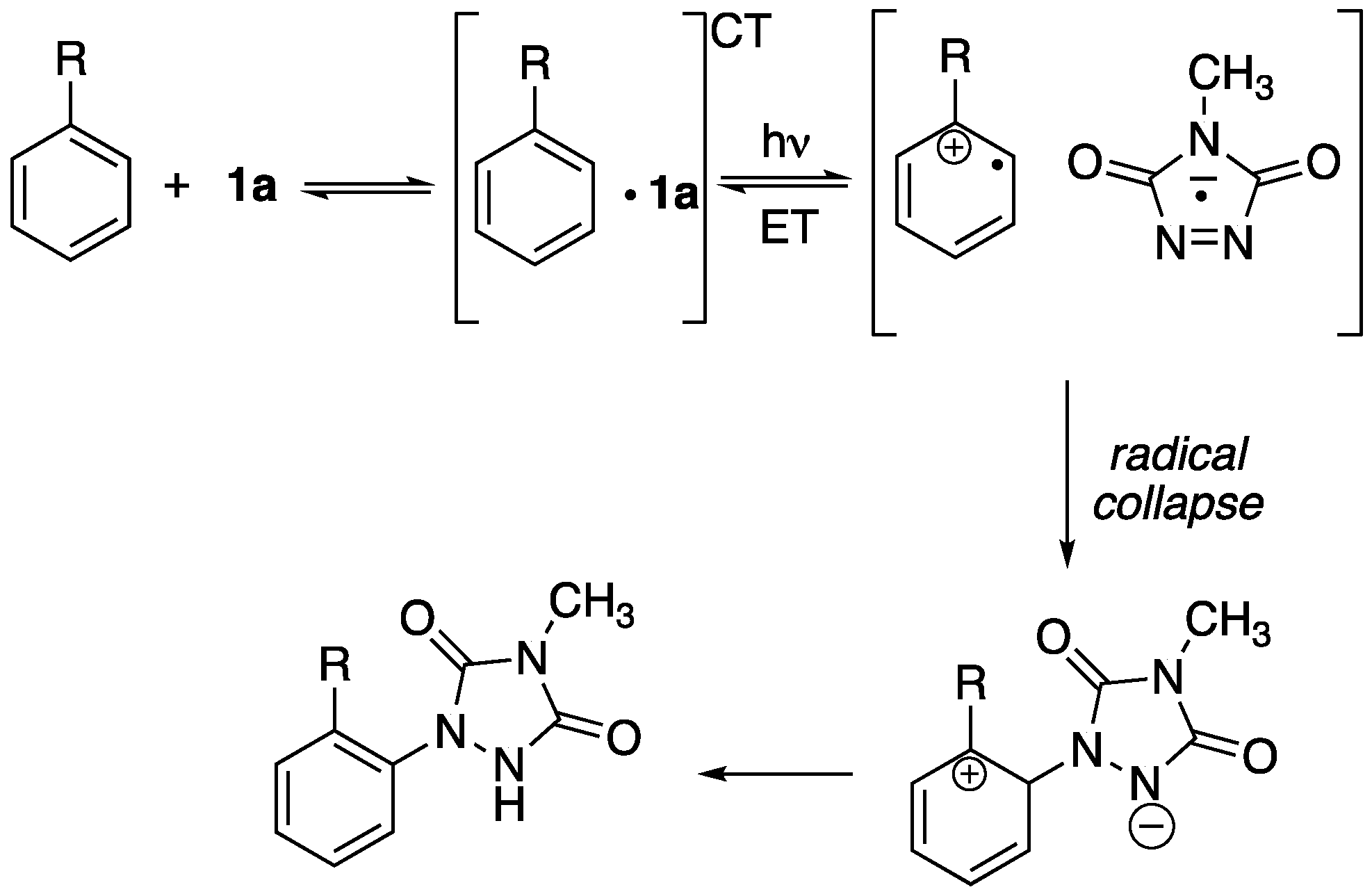
In our earlier studies on the photochemical reactions of
1a with substituted benzenes [
4], it was concluded that reaction occurred upon the irradiation of an initially formed charge transfer (CT) complex between the arene and
1a (see
Scheme 2). Irradiation promoted a single electron transfer from the benzene ring
1a, resulting in the formation of a radical cation from the aromatic substrate and a radical anion from
1a. These newly formed radicals collapsed to form a bond, thereby giving rise to the sigma complex characteristic of traditional electrophilic aromatic substitutions. Finally, proton transfer afforded the final substituted product. This electron-transfer process was promoted by the presence of electron-donating substituents on the aromatic ring. While the electron-transfer process was energetically favorable for the electron-rich substrates investigated earlier, electron transfer from unsubstituted benzene to the singlet excited state of
1a (i.e.,
11a*) is energetically disfavored. Previous estimates suggested that this process may be endothermic by as much as 15 kcal/mol [
8]. Sheridan suggested that while
11a* was certainly involved in the photochemical Diels–Alder reaction of
1a with benzene to form
4, the involvement of the corresponding triplet state (i.e.,
31a*) in this cycloaddition process remained unclear.
Chattaraj recently postulated that the “global electrophilicity index” of molecules, a measure of a compound’s willingness to react as an electrophile, is increased upon electronic excitation [
14]. Using the identical computational protocol as Chattaraj (i.e., TDDFT B3LYP/6-311+G**), a global electrophilicity index was calculated for the ground state of
1a to be 2.18 eV, a comparably equivalent electrophilicity index of 2.09 eV was calculated for the excited singlet state
11a*, and a substantially increased electrophilicity index of 3.56 eV for the excited triplet state,
31a*. Thus, electron transfer from benzene to photoexcited
1a*, while apparently not feasible from the singlet excited state, may be favorable from the triplet excited state.
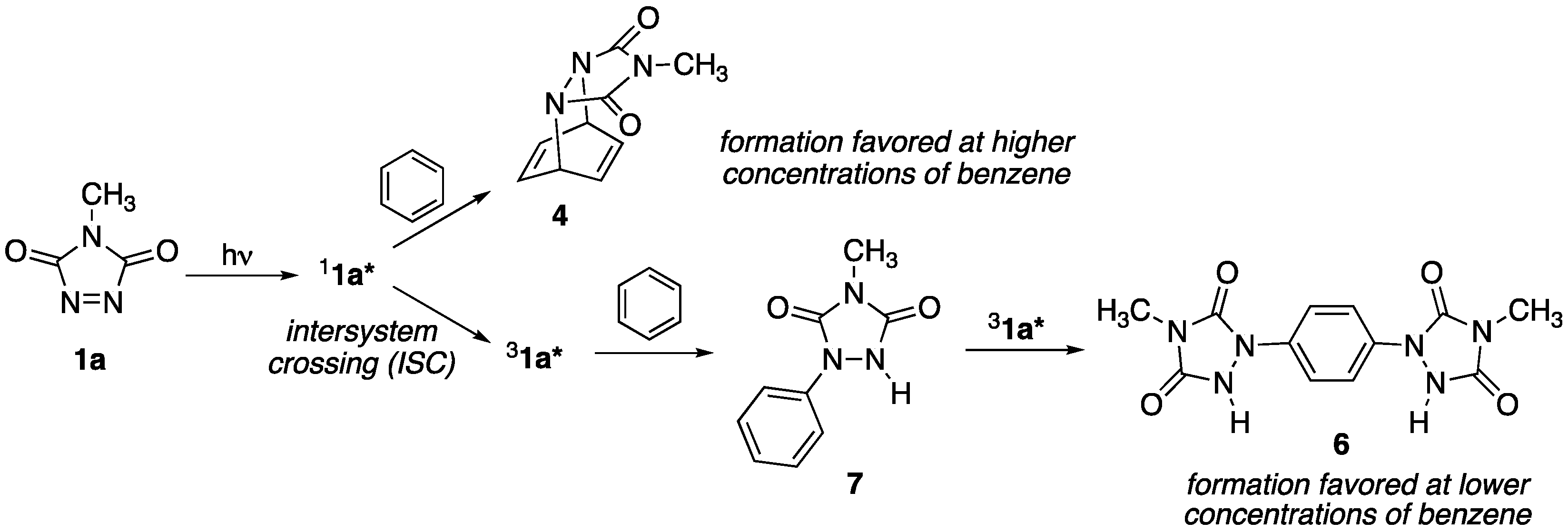
The triplet state of
1a* is populated via intersystem crossing (ISC) from the initially generated singlet state (see
Scheme 3). If, however, the singlet state is effectively quenched by reaction with benzene in a Diels–Alder cycloaddition process, there is little opportunity for the population of the triplet state. At lower concentrations of benzene, one might then expect greater opportunity for the population of the triplet state, which could then undergo reaction with benzene via an electron-transfer process. With increasing concentrations of benzene (as, for example, increasing the amount of benzene from 1 to 5 equivalents as was discussed above) one would, therefore, expect a decreased rate of reaction to form
6/
7 since more of the singlet excited state of benzene would be effectively trapped (and then replenished via the thermal retro-Diels–Alder reaction), as was experimentally observed.
Finally, electron transfer from monosubstituted 7 to 31a* to give rise to 6 should be favored relative to the electron transfer from benzene itself because the attached urazole ring acts as an electron-donating substituent, and promotes greater reactivity.
4. Conclusions
Contrary to earlier reports that suggested that there was no photochemical reaction between 1a and benzene at room temperature, it was found that they do, indeed, react. While Diels–Alder cycloadduct 4 is the product of the reaction at low temperatures (<60 °C), a bisurazole adduct 6 is the major product at room temperature. This discrepancy in observations is traced to the rapid thermal retro-Diels–Alder reaction of 4 at room temperature, which may provide an illusion of no reaction. Furthermore, while the Diels–Alder cycloaddition process takes place from the singlet state of photoactivated 1a, the electron-transfer process that leads to compounds 6/7 may take place from the triplet activated state. In the presence of higher concentrations of benzene, the cycloaddition process dominates, thereby quenching the excited state and preventing population of the triplet state 31a* via intersystem crossing. With lower concentrations of benzene, the rate of reaction via the Diels–Alder cycloaddition is decreased, and population of 31a* is made possible. Under these conditions, electron-transfer from benzene to the electrophilic 31a* occurs, leading to the formation of 7. Because the urazole ring of 7 acts as an electron-donating substituent, the rate of reaction of 7 with 31a* to form 6 is enhanced, and final yields of 6 consistently surpass those of 7.
Finally, it is of importance to note that this observed photochemical reactivity of MeTAD with benzene, and substituted benzenes [
4], conforms with some of the twelve principles previously laid out for Green Chemistry [
15]. From the standpoint of atom economy, especially when these reactions are carried out in CH
3CN as solvent during which no formation of the side-product
5 was observed, a maximum number of atoms of the starting materials are incorporated into the product. Additionally, the ability to conduct these reactions under an oxygen atmosphere at room temperature conforms to the principle of designing a reaction for energy efficiency.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}