
Photodegradation of Organic Pollutants in Seawater and Hydrogen Production via Methanol Photoreforming with Hydrated Niobium Pentoxide Catalysts


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Catalysts
2.2. Characterization of the Catalysts
2.3. Photocatalytic Tests
2.3.1. Photodegradation of Phenol in Seawater
- (a)
- Photolysis: 25 mL of phenolic solution, without catalyst, under UV radiation (253.7 nm) for 120 min.
- (b)
- Adsorption: 25 mL of phenolic solution, catalyst dosage of 0.5 gcat·L−1, in the absence of UV radiation (in dark) for 120 min.
- (c)
- Heterogeneous photocatalysis under UV radiation: 25 mL of phenolic solution, under UV radiation (253.7 nm). In this case, the following parameters were varied: catalyst dosage (0.5–2 gcat·L−1) and time of irradiation under UV light (0–120 min).
- (d)
- Heterogeneous photocatalysis under UV radiation using Pt-promoted catalysts: For this test, Pt (1, 5, and 10% w/w) was loaded on the surface of each catalyst by an in situ photodeposition method by using aqueous H2PtCl6 solution as the Pt source. In this case, the photocatalytic tests were carried out under the optimal conditions determined in experiments described in items a–c.
2.3.2. Reuse of Catalysts
2.4. Hydrogen Production by Photoreforming of Methanol
2.5. Hydroxyl Radical (•OH) Measuring Experiments
3. Results and Discussion
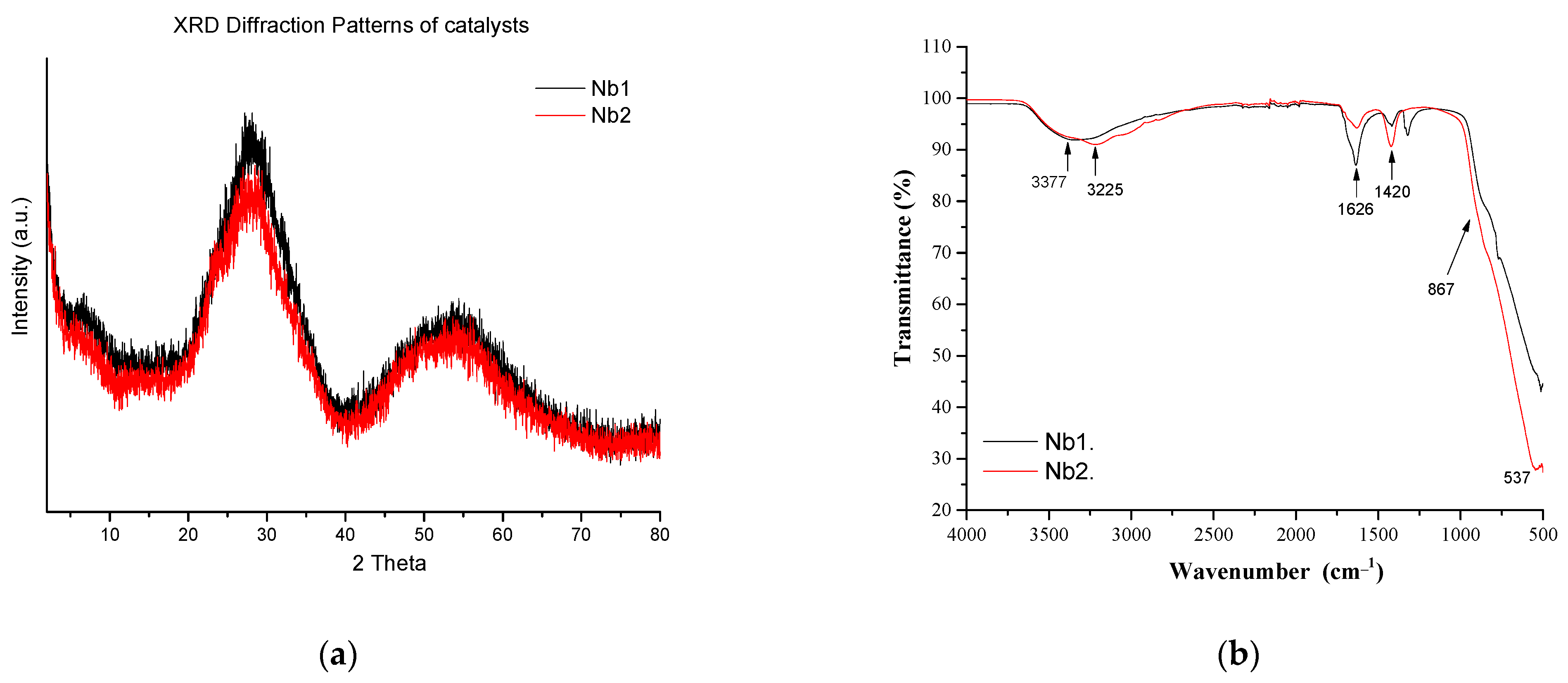
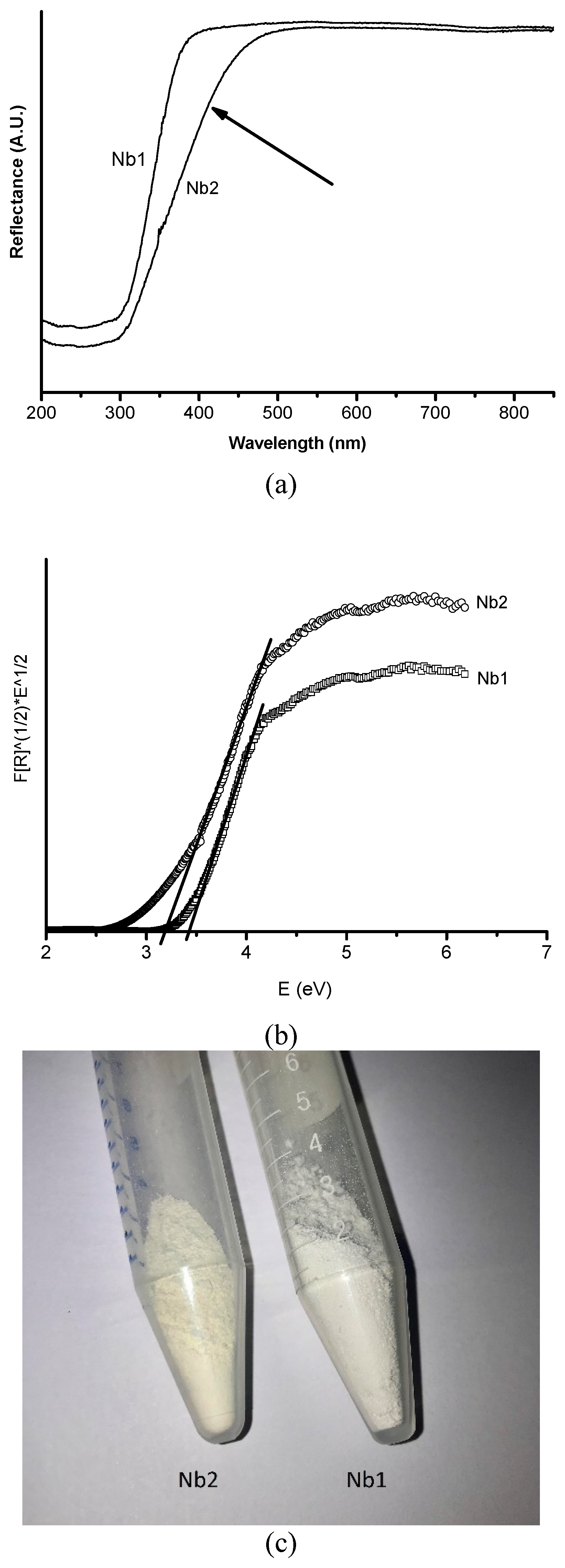
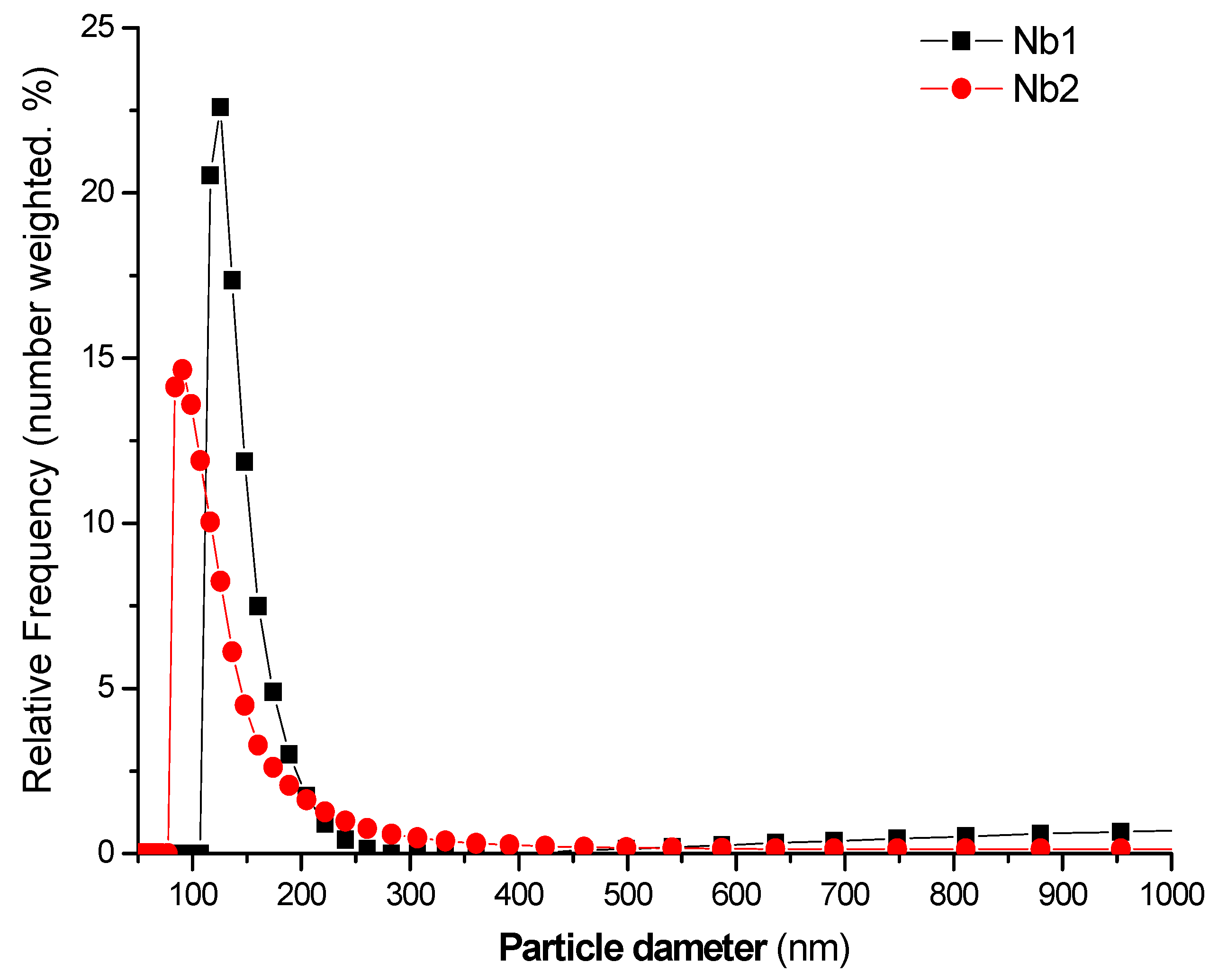
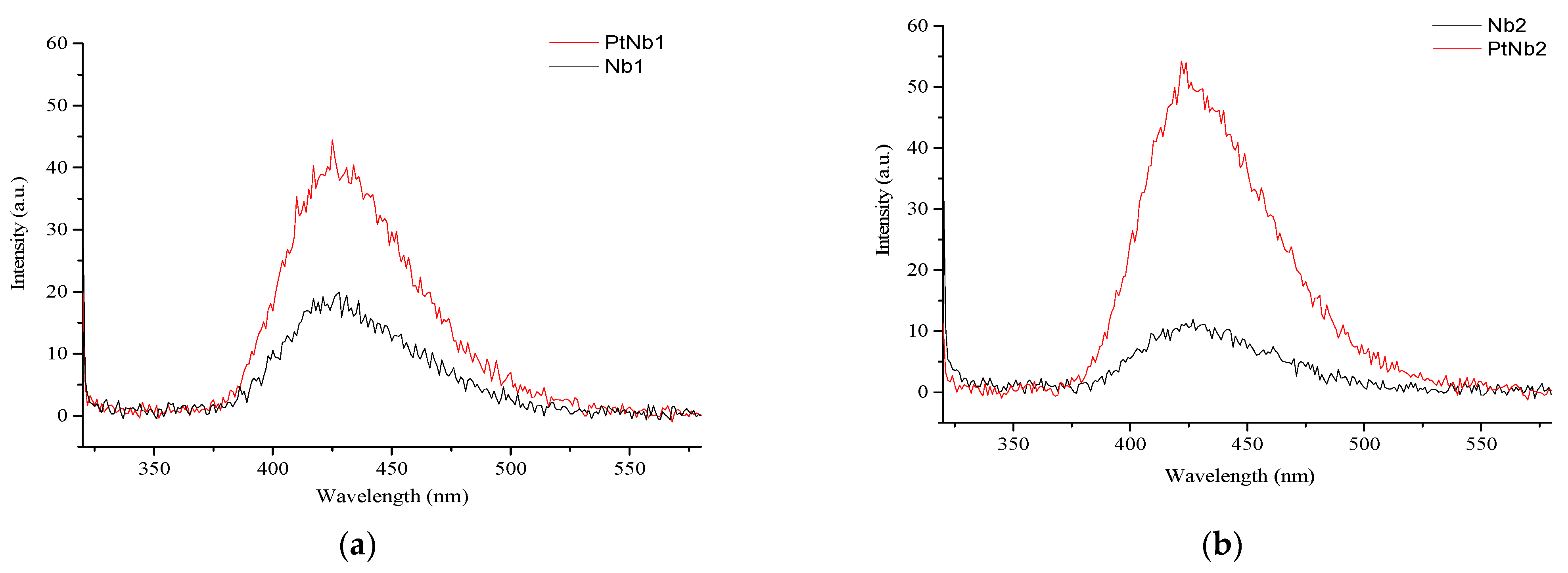
3.1. Characterization of the Catalysts
3.2. Photocatalytic Tests
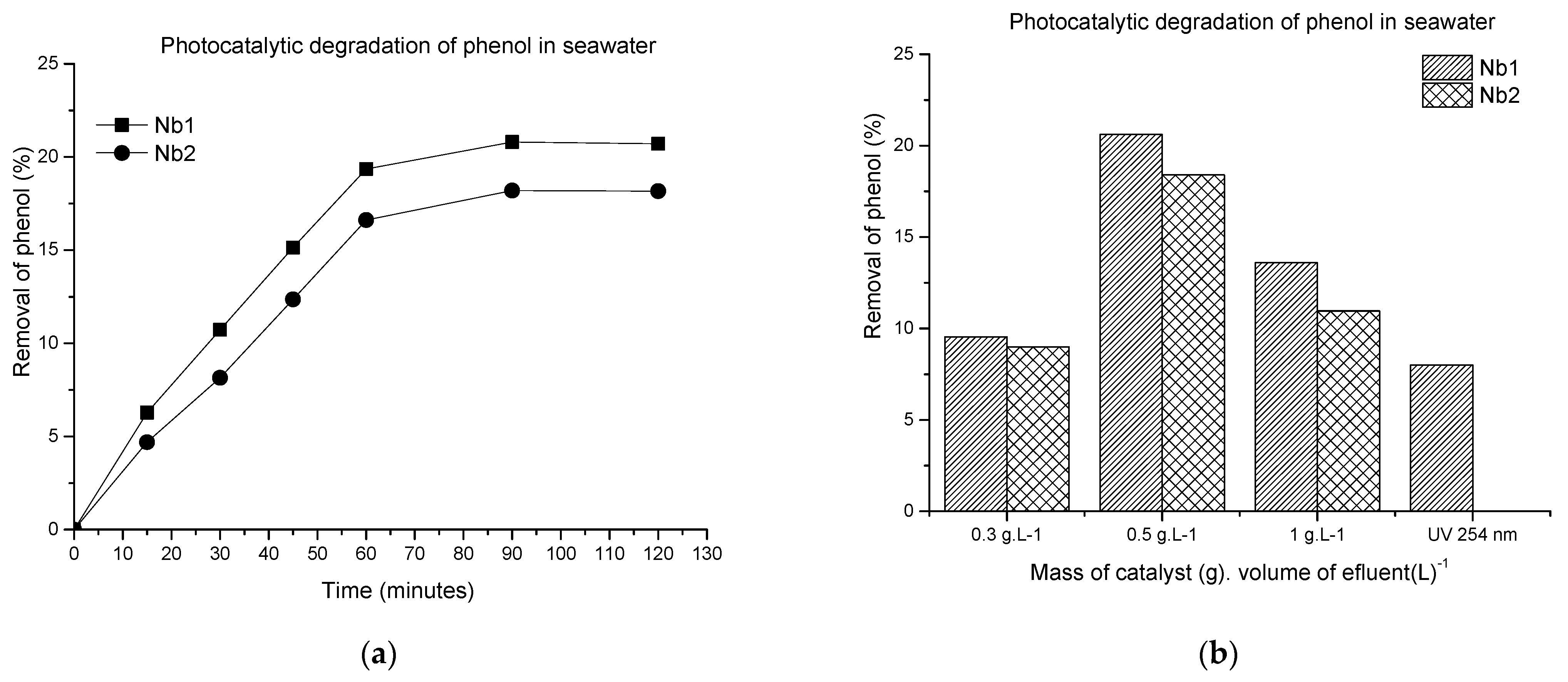
3.2.1. Photocatalytic Degradation of Phenol in Seawater
Variation in Irradiation Time and Catalyst Dosage
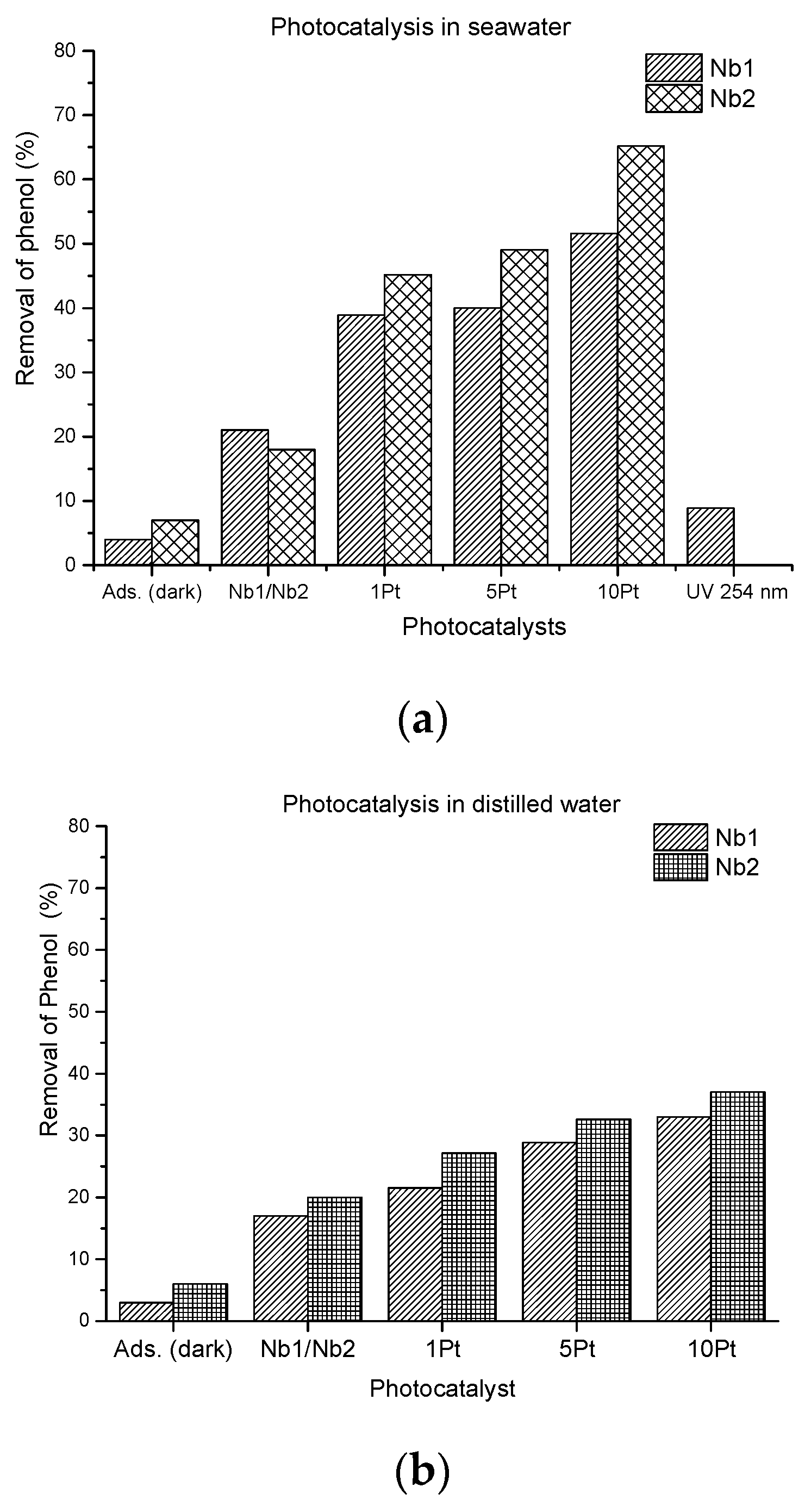
Comparison of Photocatalysis in Seawater and Distilled Water
Presence of Pt° Promoter
Possible Mechanism
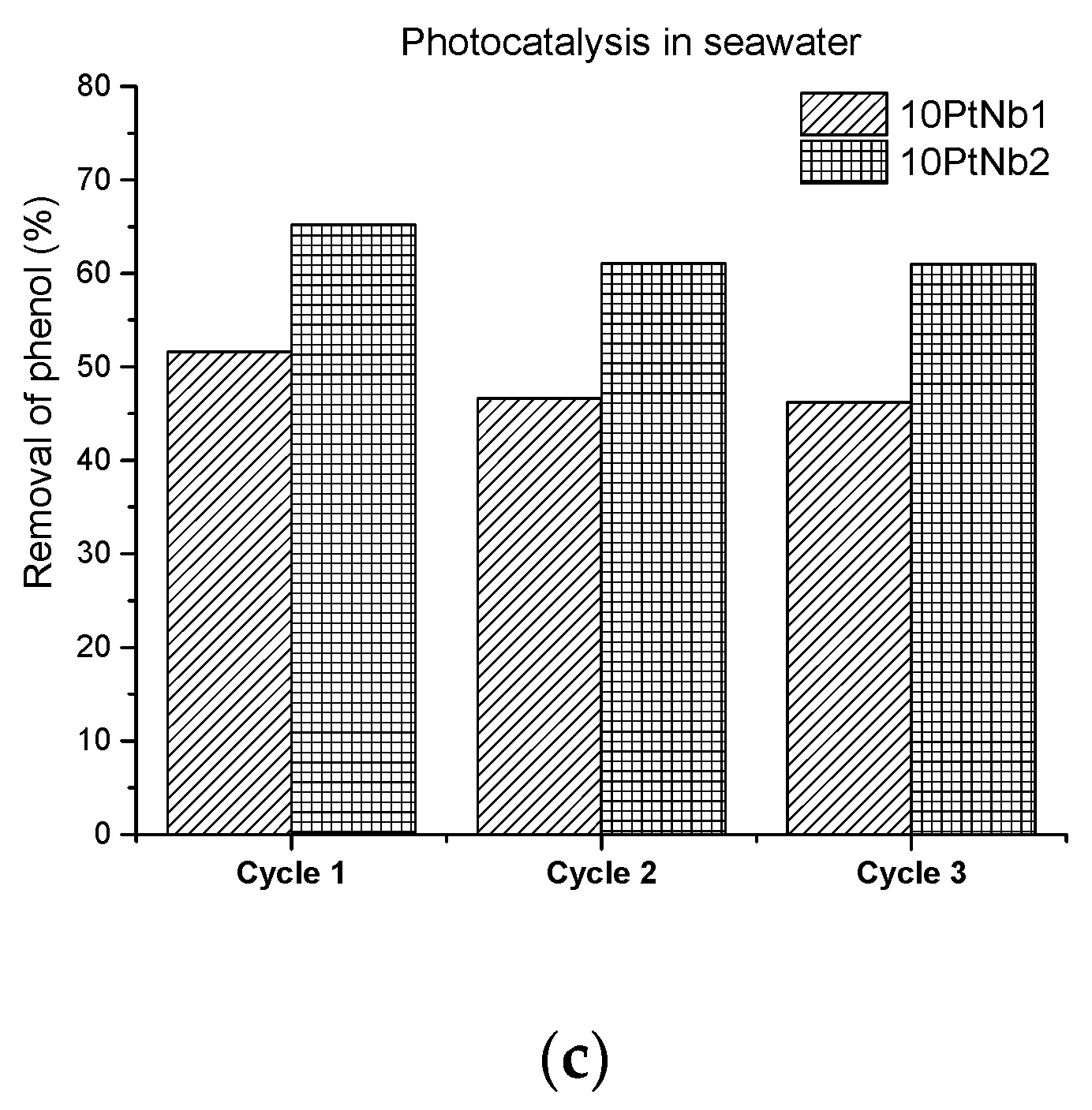
Catalyst Reuse
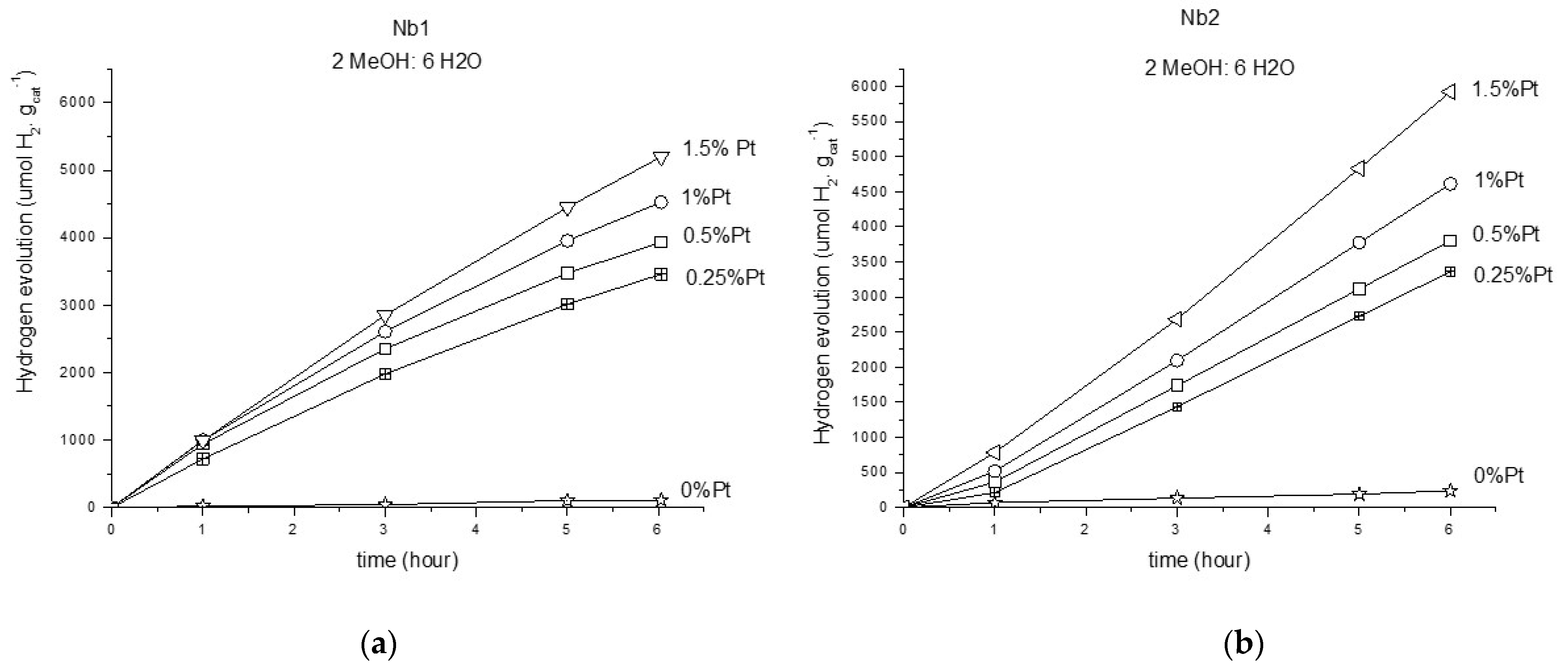
3.2.2. Photoreforming of Methanol
Possible Mechanism
Hydroxyl Radical (•OH) Measuring Experiments
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakagomi, F.; Cerruti, S.E.; Freitas, M.R.; Neto, E.S.F.; Andrade, F.V.; Siqueira, G.O. Niobium pentoxide produced by a novel method microwave assisted combustion synthesis. Chem. Phys. Lett. 2019, 729, 37–41. [Google Scholar] [CrossRef]
- Oliveira, H.S.; Resende, J.M.; Souza, P.P.; Patrício, P.S.O.; Oliveira, L.C.A. Synthetic Niobium Oxyhydroxide as a Bifunctional Catalyst for Production of Ethers and Allyl Alcohol from Waste Glycerol. J. Braz. Chem. Soc. 2017, 28, 2244–2253. [Google Scholar] [CrossRef]
- Padula, I.D.; Chagas, P.; Furst, C.G.; Oliveira, L.C.A. Mesoporous Niobium Oxyhydroxide Catalysts for Cyclohexene Epoxidation Reactions. Appl. Sci. 2018, 8, 881. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, N.P.; Nogueira, A.E.; Marcos, F.C.F.; Machado, V.A.; Rocca, R.R.; Assaf, E.M.; Asencios, Y.J.O. CeO2–Nb2O5 photocatalysts for degradation of organic pollutants in water. Rare Met. 2020, 39, 230–240. [Google Scholar] [CrossRef]
- Ferraz, N.P.; Marcos, F.C.F.; Nogueira, A.E.; Martins, A.S.; Lanza, M.R.V.; Assaf, E.M.; Asencios, Y.J.O. Hexagonal-Nb2O5/Anatase-TiO2 mixtures and their applications in the removal of Methylene Blue dye under various conditions. Mater. Chem. Phys. 2017, 198, 331–340. [Google Scholar] [CrossRef]
- Asencios, Y.J.O.; Lourenço, V.S.; Carvalho, W.A. Removal of phenol in seawater by heterogeneous photocatalysis using activated carbon materials modified with TiO2. Catal. Today 2020, 388–389, 247–258. [Google Scholar] [CrossRef]
- Nowak, I.; Ziolek, M. Niobium Compounds: Preparation, Characterization, and Application in Heterogeneous Catalysis. Chem. Rev. 1999, 99, 3603–3624. [Google Scholar] [CrossRef]
- Ziolek, M.; Sobczak, I.; Decyk, P.; Sobanska, K.; Pietrzyk, P.; Sojka, Z. Search for reactive intermediates in catalytic oxidation with hydrogen peroxide over amorphous niobium(V) and tantalum(V) oxides. Appl. Catal. B 2015, 164, 288–296. [Google Scholar] [CrossRef]
- Iizuka, T.; Ogasawara, K.; Tanabe, K. Acidic and Catalytic Properties of Niobium Pentaoxide. Bull. Chem. Soc. Jpn. 1983, 56, 2927–2931. [Google Scholar] [CrossRef]
- Murayama, T.; Chen, J.; Hirata, J.; Matsumoto, K.; Ueda, W. Hydrothermal synthesis of octahedra-based layered niobium oxide and its catalytic activity as a solid acid. Catal. Sci. Technol. 2014, 4, 4250–4257. [Google Scholar] [CrossRef]
- Oliveira, L.C.A.; Costa, N.T.; Pliego, J.R.; Silva, A.C.; de Souza, P.P.; Patrícia, P.S. Amphiphilic niobium oxyhydroxide as a hybrid catalyst for sulfur removal from fuel in a biphasic system. Appl. Catal. B Environ. 2014, 147, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Ekhsan, J.M.; Lee, S.L.; Nur, H. Niobium oxide and phosphoric acid impregnated silica–titania as oxidative-acidic bifunctional catalyst. Appl. Catal. A Gen. 2014, 471, 142–148. [Google Scholar] [CrossRef]
- Da Silva, G.T.; Nogueira, A.E.; Oliveira, J.A.; Torres, J.A.; Lopes, O.F.; Ribeiro, C. Acidic surface niobium pentoxide is catalytic active for CO2 photoreduction. Appl. Catal. B Environ. 2019, 242, 349–357. [Google Scholar] [CrossRef]
- Moraes, N.P.; Pinto da Silva, M.L.C.; Rodrigues, L.A. Effect of metal doping in the photocatalytic properties of carbon xerogel-Nb2O5 composite towards visible light degradation of methylene blue. Mater. Lett. 2018, 228, 486–489. [Google Scholar] [CrossRef]
- Feldmann, C.; Jungk, H.O. Polyol-Mediated Preparation of Nanoscale Oxide Particles. Angew. Chem. Int. Ed. 2001, 40, 359–362. [Google Scholar] [CrossRef]
- Uekawa, N.; Kudo, T.; Mori, F.; Jun Wu, Y.; Kakegawa, K. Low-temperature synthesis of niobium oxide nanoparticles from peroxo niobic acid sol. J. Colloid Interface Sci. 2003, 264, 378–384. [Google Scholar] [CrossRef]
- Leite, E.R.; Vila, C.; Bettini, J.; Longo, E. Synthesis of Niobia Nanocrystals with Controlled Morphology. J. Phys. Chem. B 2006, 37, 18088–18090. [Google Scholar] [CrossRef]
- Mozetič, M.; Cvelbar, U.; Sunkara, M.K.; Vaddiraju, S. A Method for the Rapid Synthesis of Large Quantities of Metal Oxide Nanowires at Low Temperatures. Adv. Mater. 2005, 17, 2138–2142. [Google Scholar] [CrossRef]
- Hu, W.; Liu, Z.; Nie, G.; Mi, Y.; Zhao, Y.; Yao, K. Synthesis of Nb3.49N4.56O0.44 nanoplatelets and NbS2–Nb2O5 nanoflakes. Mater. Chem. Phys. 2009, 113, 511–514. [Google Scholar] [CrossRef]
- Pinna, N.; Antonietti, M.; Niederberger, M. A novel non-aqueous route to V2O3 and Nb2O5 nanocrystals. Colloids Surf. A 2004, 250, 211–213. [Google Scholar] [CrossRef]
- Carvalho, K.T.G.; Silva, A.C.; Oliveira, L.C.A.; Gonçalves, M.; Magriotis, Z.M. Nióbia sintética modificada como catalisador na oxidação de corante orgânico: Utilização de H2O2 e O2 atmosférico como oxidantes. Quím. Nova 2009, 32, 1373–1377. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.L.; Tauc, J. Weak Absorption Tails in Amorphous Semiconductors. Phys. Rev. B 1972, 5, 3144. [Google Scholar] [CrossRef]
- Carpio, E.; Zúñiga, P.; Ponce, S.; Solis, J.; Rodriguez, J.; Estrada, W. Photocatalytic degradation of phenol using TiO2 nanocrystals supported on activated carbon. J. Mol. Catal. A Chem. 2005, 228, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.S. Integration of Flotation and Photo-Fenton Processes to Reduce Oils and Greases from Water Produced in Oil Fields. Ph.D. Thesis, Federal University of Rio Grande do Norte (UFRN), Natal, Brazil, 2013. Available online: https://repositorio.ufrn.br/jspui/handle/123456789/15924 (accessed on 9 April 2022).
- EPA (U.S. Environmental Protection Agency). Method 604, Methods for Organic Chemical Analysis of Municipal and Industrial Wastewater; Part VIII, 40 CFR Part 136. Appendix A; Environmental Protection Agency EPA: Washington, DC, USA, 1984.
- APHA Method D 5530. Standard Methods for the Examination of Water and Wastewater, 17th ed.; American Public Health Association (APHA): Washington, DC, USA, 1989. [Google Scholar]
- Sun, X.; Zhang, H.; Wei, J.; Yu, Q.; Yang, P.; Zhang, F. Preparation of point-line Bi2WO6@TiO2 nanowires composite photocatalysts with enhanced UV/visible-light-driven photocatalytic activity. Mater. Sci. Semicond. Process. 2016, 45, 51–56. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, L.; Jianga, B.; Zheng, J.; Hu, P.; Li, S.; Wu, M.; Wu, W. Fabrication of Z-scheme Ag3PO4/MoS2 composites with enhanced photocatalytic activity and stability for organic pollutant degradation. Appl. Surf. Sci. 2016, 377, 99–108. [Google Scholar] [CrossRef]
- Xu, D.; Cheng, B.; Cao, S.; Yu, J. Enhanced photocatalytic activity and stability of Z-scheme Ag2CrO4-GO composite photocatalysts for organic pollutant degradation. Appl. Catal. B 2015, 164, 380–388. [Google Scholar] [CrossRef]
- Tong, T.; Zhang, H.; Chen, J.; Jin, D.; Cheng, J. The photocatalysis of BiFeO3 disks under visible light irradiation. Catal. Commun. 2016, 87, 23–26. [Google Scholar] [CrossRef]
- Rosli, N.I.M.; Lam, S.-M.; Sin, J.C.; Satoshi, I.; Mohamed, A.R. Photocatalytic Performance of ZnO/g-C3N4 for Removal of Phenol under Simulated Sunlight Irradiation. J. Environ. Eng. 2018, 144, 04017091. [Google Scholar] [CrossRef]
- Luis Marin, M.; Hallett-Tapley, G.L.; Impellizzeri, S.; Fasciani, C.; Simoncelli, S.; Netto-Ferreira, J.C.; Scaiano, J.C. Synthesis, acid properties and catalysis by niobium oxide nanostructured materials. Catal. Sci. Technol. 2014, 4, 3044–3052. [Google Scholar] [CrossRef] [Green Version]
- Brandão, R.F.; Quirino, R.L.; Mello, V.M.; Tavares, A.P.; Peres, A.C.; Guinhos, F.; Rubim, J.C.; Suarez, P.A.Z. Synthesis, Characterization and use of Nb2O5 based Catalysts in Producing Biofuels by Transesterification, Esterification and Pyrolysis. J. Braz. Chem. Soc. 2009, 20, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, F.C.; Carvalho, W.M.; Oliveira, L.C.A.; Passos, A.R.; Silva, A.C. Utilization of Sn/Nb2O5 composite for removal of Methylene blue. Quim. Nova 2010, 33, 528–531. [Google Scholar]
- Saranya, K.; Raja, S.T.K.; Subhasree, R.S.; Gnanamani, A.; Das, S.K.; Rajendran, N. Fabrication of nanoporous sodium niobate coating on 316L SS for Orthopaedics. Ceram. Int. 2017, 43, 11569–11579. [Google Scholar] [CrossRef]
- Clechet, P.; Martin, J.R.; Olier, R.; Vallouy, C. Photoeletrochemical effect on several transition metal oxides. C. R. Acad. Sci. Ser. C 1976, 282, 887. [Google Scholar]
- Brayner, R.; Bozon-Verduraz, F. Niobium pentoxide prepared by soft chemical routes: Morphology, structure, defects and quantum size effect. Phys. Chem. Chem. Phys. 2003, 5, 1457–1466. [Google Scholar] [CrossRef]
- Nogueira, A.E.; Ramalho, T.C.; Oliveira, L.C.A. Photocatalytic Degradation of Organic Compound in Water Using Synthetic Niobia: Experimental and Theoretical Studies. Top. Catal. 2011, 54, 270–276. [Google Scholar] [CrossRef]
- Iguchi, S.; Teramura, K.; Hosokawa, S.; Tanaka, T. Effect of chloride ion as a hole scavenger on the photocatalytic conversion of CO2 in an aqueous solution over Ni−Al layered double hydroxides. Phys. Chem. Chem. Phys. 2015, 17, 17995–18003. [Google Scholar] [CrossRef] [Green Version]
- Hippargi, G.; Mangrulkar, P.; Chilkalwar, A.; Labhsetwar, N.; Rayalu, S. Chloride ion: A promising hole scavenger for photocatalytic hydrogen generation. Int. J. Hydrogen Energy 2018, 43, 6815–6823. [Google Scholar] [CrossRef]
- Demianov, P.; Concetta, D.E.; Giancuzza, A.S.; Ammartano, S. Equilibrium studies in natural waters: Speciation of phenolic compounds in synthetic seawater at different salinities. Environ. Toxicol. Chem. 1995, 5, 767–773. [Google Scholar] [CrossRef]
- Patnaik, P.; Khoury, J.N. Reaction of phenol with nitrite ion: Pathways of formation of nitrophenols in environmental waters. Water Res. 2004, 38, 206–210. [Google Scholar] [CrossRef]
- Moussavi, M. Effect of polar substituents on autoxidation of phenols. Water Res. 1979, 13, 1125–1128. [Google Scholar] [CrossRef]
- Kinney, L.C.; Ivanuski, V.R. Water Research Center. Advance Waste Treatment Research Laboratory-XIII., Photolysis Mechanisms for Pollution Abatement; U.S. Department of the Interior. Federal Water Pollution Control Administration: Cincinnati, OH, USA, 1969. Available online: https://www.epa.gov/nscep (accessed on 9 April 2022).
- Idrees, F.; Dillert, R.; Bahnemann, D.; Butt, F.K.; Tahir, M. In-Situ Synthesis of Nb2O5/g-C3N4 Heterostructures as Highly Efficient Photocatalysts for Molecular H2 Evolution under Solar Illumination. Catalysts 2019, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yang, H.-C.; Wang, W.-L. Synthesis of mesoporous Nb2O5 photocatalysts with Pt, Au, Cu and NiO cocatalyst for water splitting. Catal. Today 2011, 174, 106–113. [Google Scholar] [CrossRef]
- Yao, S.; Li, J.; Shi, Z. Immobilization of TiO2 nanoparticles on activated carbon fiber and its photodegradation performance for organic pollutants. Particuology 2010, 8, 272–278. [Google Scholar] [CrossRef]
- Ziolli, R.L.; Jardim, W.F. Photocatalytic decomposition of seawater-soluble crude-oil fractions using high surface area colloid nanoparticles of TiO2. J. Photochem. Photobiol. A Chem. 2002, 147, 205–212. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Nomikos, G.N.; Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Kinetic and mechanistic study of the photocatalytic reforming of methanol over Pt/TiO2 catalyst. Appl. Catal. B Environ. 2014, 146, 249–257. [Google Scholar] [CrossRef]
- Yu, Z.; Chuang, S.S.C. In situ IR study of adsorbed species and photogenerated electrons during photocatalytic oxidation of ethanol on TiO2. J. Catal. 2007, 246, 118–126. [Google Scholar] [CrossRef]
- Yu, J.; Wang, G.; Cheng, B.; Zhou, M. Effects of hydrothermal temperature and time on the photocatalytic activity and microstructures of bimodal mesoporous TiO2 powders. Appl. Catal. B Environ. 2007, 69, 171–180. [Google Scholar] [CrossRef]
- Puangpetch, T.; Sreethawong, T.; Yoshikawa, S.; Chavadej, S. Hydrogen production from photocatalytic water splitting over mesoporous-assembled SrTiO3 nanocrystal-based photocatalysts. J. Mol. Catal. A Chem. 2009, 312, 97–106. [Google Scholar] [CrossRef]
- Lin, H.Y.; Huang, H.C.; Wang, W.L. Preparation of mesoporous In–Nb mixed oxides and its application in photocatalytic water splitting for hydrogen production. Microporous Mesoporous Mater. 2008, 115, 568–575. [Google Scholar] [CrossRef]
- Won, D.; Hyun, H.; Jum, G.K.; Sang, S.J.; Sang, W.B.; Jae, M.J.; Lee, S. Photocatalytic decomposition of water–methanol solution over metal-doped layered perovskites under visible light irradiation. Catal. Today 2004, 93–95, 845–850. [Google Scholar]
- Xu, D.; Li, L.; Xia, T.; Fan, W.; Wang, F.; Bai, H.; Shi, W. Heterojunction composites of g-C3N4/KNbO3 enhanced photocatalytic properties for water splitting. Int. J. Hydrogen Energy 2018, 43, 16566–16572. [Google Scholar] [CrossRef]
- Chen, T.; Feng, Z.; Wu, G.; Shi, J.; Ma, G.; Ying, P.; Li, C.J. Mechanistic Studies of Photocatalytic Reaction of Methanol for Hydrogen Production on Pt/TiO2 by in situ Fourier Transform IR and Time-Resolved IR Spectroscopy. J. Phys. Chem. C 2007, 111, 8005–8014. [Google Scholar] [CrossRef]
- Kecskés, T.; Raskó, J.; Kiss, J. FTIR and mass spectrometric studies on the interaction of formaldehyde with TiO2 supported Pt and Au catalysts. Appl. Catal. A 2004, 273, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Granone, L.I.; Sieland, F.; Zheng, N.; Dillert, R.; Bahnemann, D.W. Photocatalytic conversion of biomass into valuable products: A meaningful approach? Green Chem. 2018, 20, 1169–1192. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W% (EDX) | BET Surface Area | Zeta Potential | Band Gap Energy | |||||
|---|---|---|---|---|---|---|---|---|
| O | Na | Cl | K | Nb | (m2·g−1) | (mV) | (eV) | |
| Nb1 | 75.5 | 2.7 | 0.7 | 0.4 | 20.7 | 14.0 | −41.4 | 3.5 |
| Nb2 | 76.1 | 2.3 | 0.6 | 0.3 | 20.7 | 22.8 | −45.7 | 3.2 |
| Catalyst | Solution | Volume Ratio | Temperature °C | H2 Production (µmol·h−1·g−1) | References |
|---|---|---|---|---|---|
| Nb1 | MeOH: H2O | 2:6 | room temp. | 18.5 | This work |
| 1%Pt Nb1 | MeOH: H2O | 2:6 | room temp. | 754 | This work |
| 1.5%Pt Nb1 | MeOH: H2O | 2:6 | room temp. | 866 | This work |
| Nb2 | MeOH: H2O | 2:6 | room temp. | 40 | This work |
| 1%Pt Nb2 | MeOH: H2O | 2:6 | room temp. | 769 | This work |
| 1.5%Pt Nb2 | MeOH: H2O | 2:6 | room temp. | 988 | This work |
| 0.5%Pt/SrTiO3 | MeOH: H2O | 2:8 | 15 | ≈210 | Puangpetch et al. [54] |
| Nb2O5 (mesoporous) | MeOH: H2O | 1:5 | 42 | 58.2 | Lin et al. [55] |
| 1%Pt Nb2O5 | MeOH: H2O | 1:5 | 43 | 4647 | Lin et al. [47] |
| TiO2 (P25) | MeOH: H2O | 1:5 | 43 | 100.7 | Lin et al. [47] |
| 1%Pt/Cr-La2Ti2O7 | MeOH: H2O | 1:2 | - | 30 | Won et al. [56] |
| 1:1/g-CN:KNO graphitic carbon nitride/KNbO3 | Triethanolamine: H2O | 2:8 | 5 | 19.95 | Xu et al. [57] |
| 2%Pt 1:1/CN:KNO | Triethanolamine: H2O | 2:8 | 5 | 101 9.38 | Xu et al. [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asencios, Y.J.O.; Machado, V.A. Photodegradation of Organic Pollutants in Seawater and Hydrogen Production via Methanol Photoreforming with Hydrated Niobium Pentoxide Catalysts. Sustain. Chem. 2022, 3, 172-191. https://doi.org/10.3390/suschem3020012
Asencios YJO, Machado VA. Photodegradation of Organic Pollutants in Seawater and Hydrogen Production via Methanol Photoreforming with Hydrated Niobium Pentoxide Catalysts. Sustainable Chemistry. 2022; 3(2):172-191. https://doi.org/10.3390/suschem3020012
Chicago/Turabian StyleAsencios, Yvan J. O., and Vanessa A. Machado. 2022. "Photodegradation of Organic Pollutants in Seawater and Hydrogen Production via Methanol Photoreforming with Hydrated Niobium Pentoxide Catalysts" Sustainable Chemistry 3, no. 2: 172-191. https://doi.org/10.3390/suschem3020012
APA StyleAsencios, Y. J. O., & Machado, V. A. (2022). Photodegradation of Organic Pollutants in Seawater and Hydrogen Production via Methanol Photoreforming with Hydrated Niobium Pentoxide Catalysts. Sustainable Chemistry, 3(2), 172-191. https://doi.org/10.3390/suschem3020012