
Does Dementia Have a Microbial Cause?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
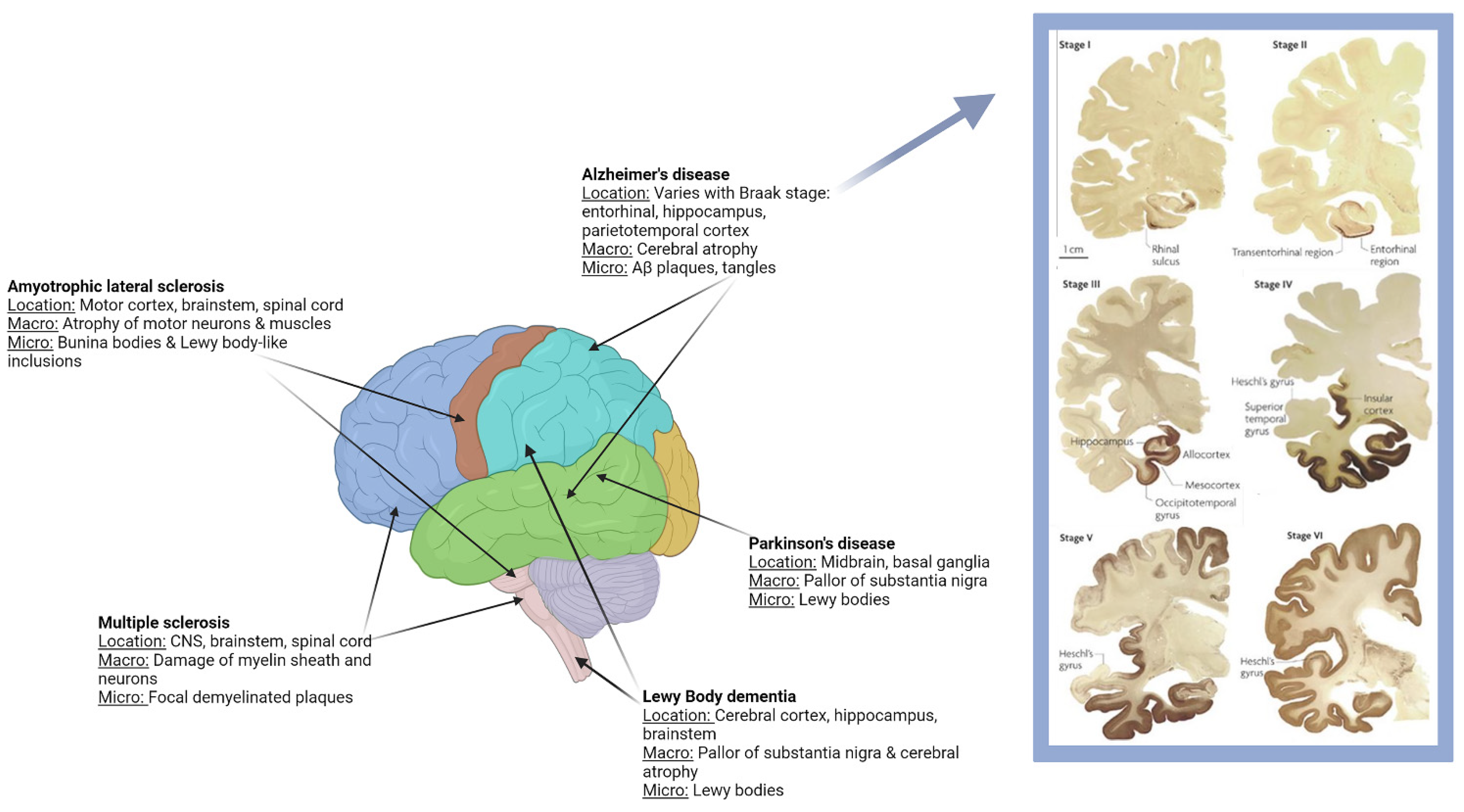
1.1. Clinical and Pathological Characteristics
1.2. Pathogenesis of Alzheimer’s Disease and Associated Dementias
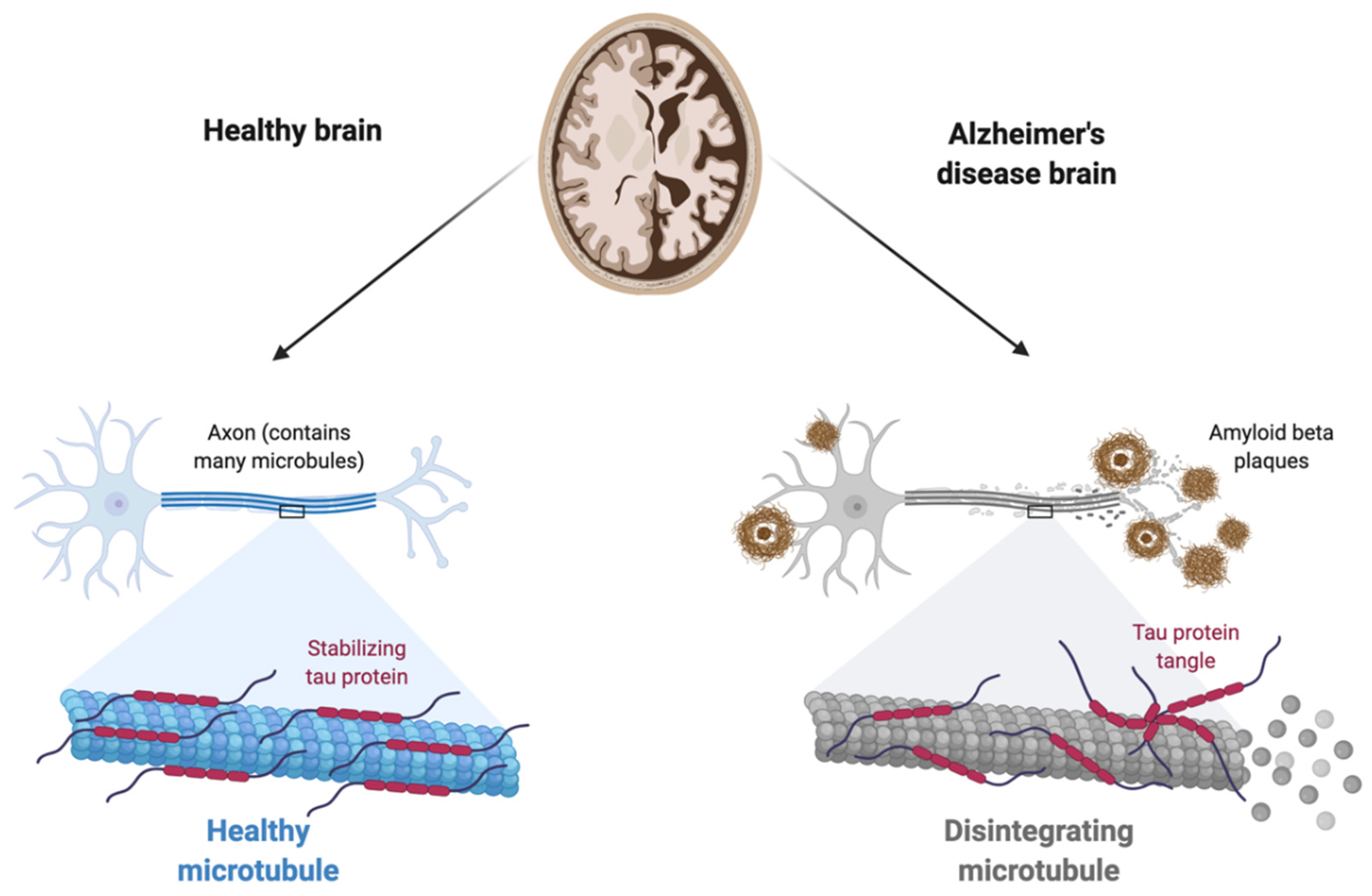
1.2.1. The Amyloid Hypothesis
1.2.2. The Inflammatory Hypothesis
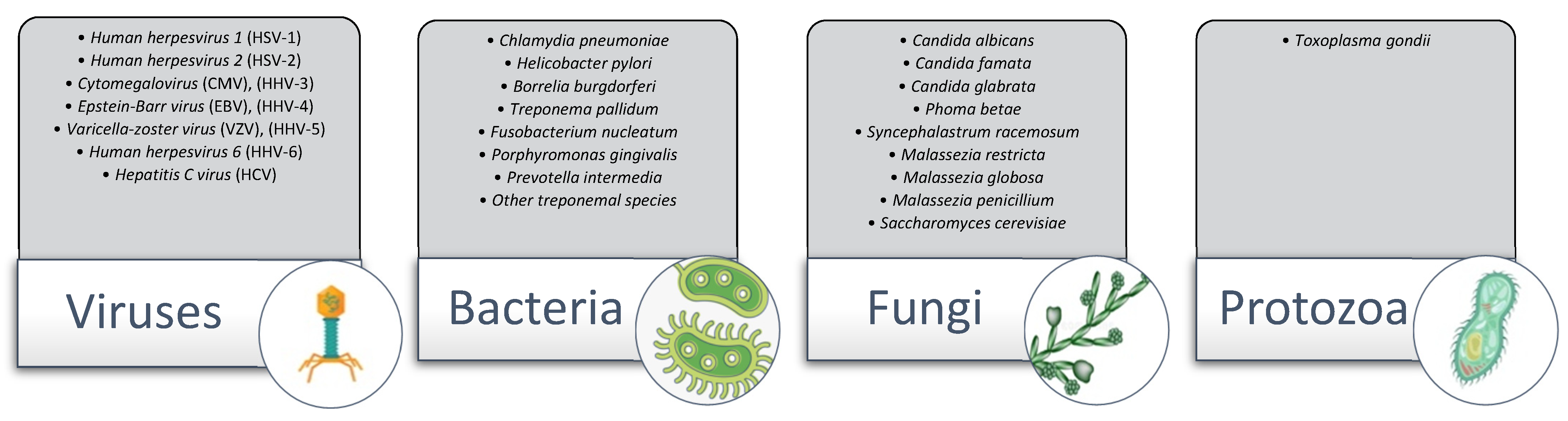
1.2.3. The Microbial Hypothesis
1.2.4. Role of the Gut–Brain Axis (GBA)
2. Discussion: The Microbial Hypothesis
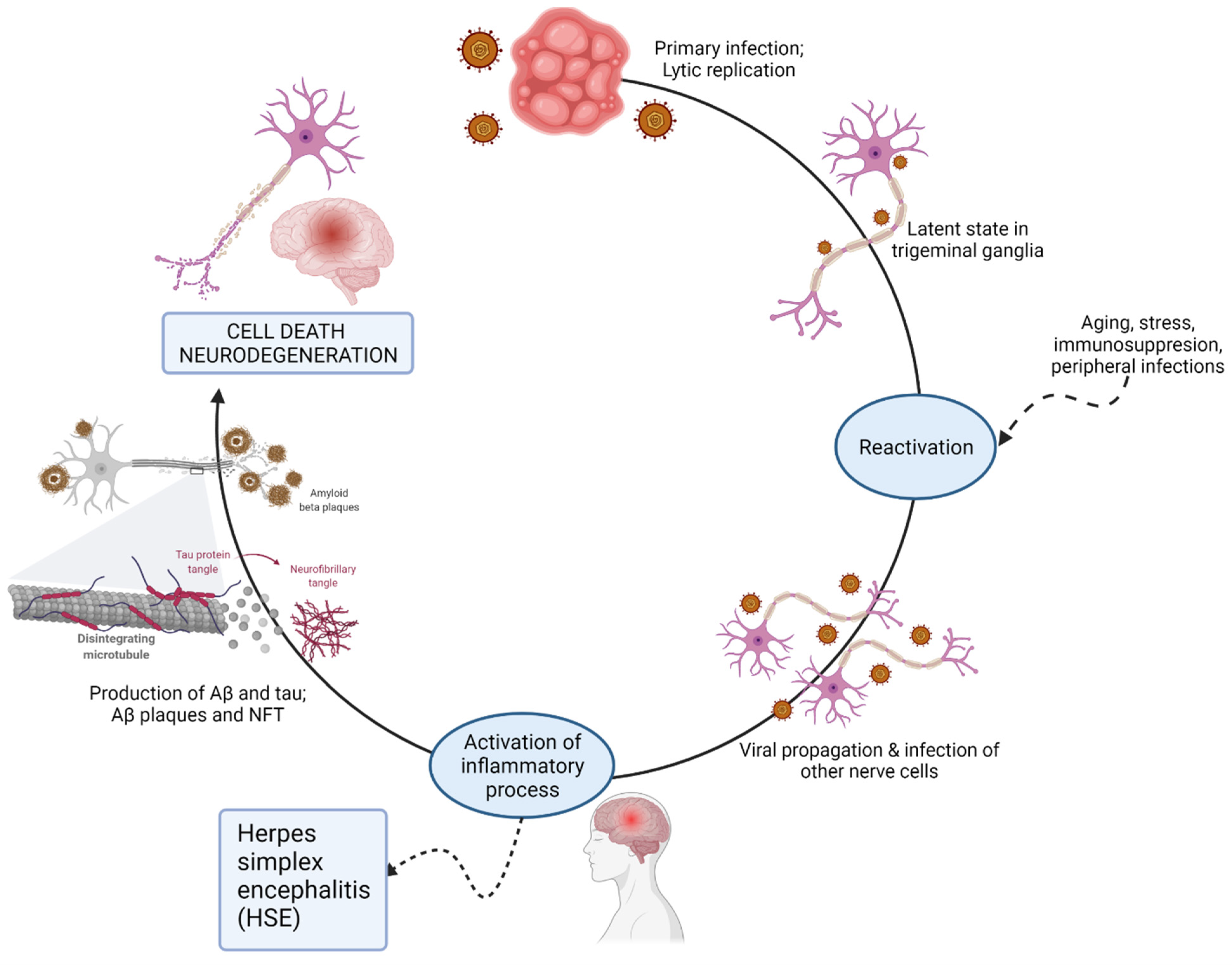
2.1. The Herpesviridae Family
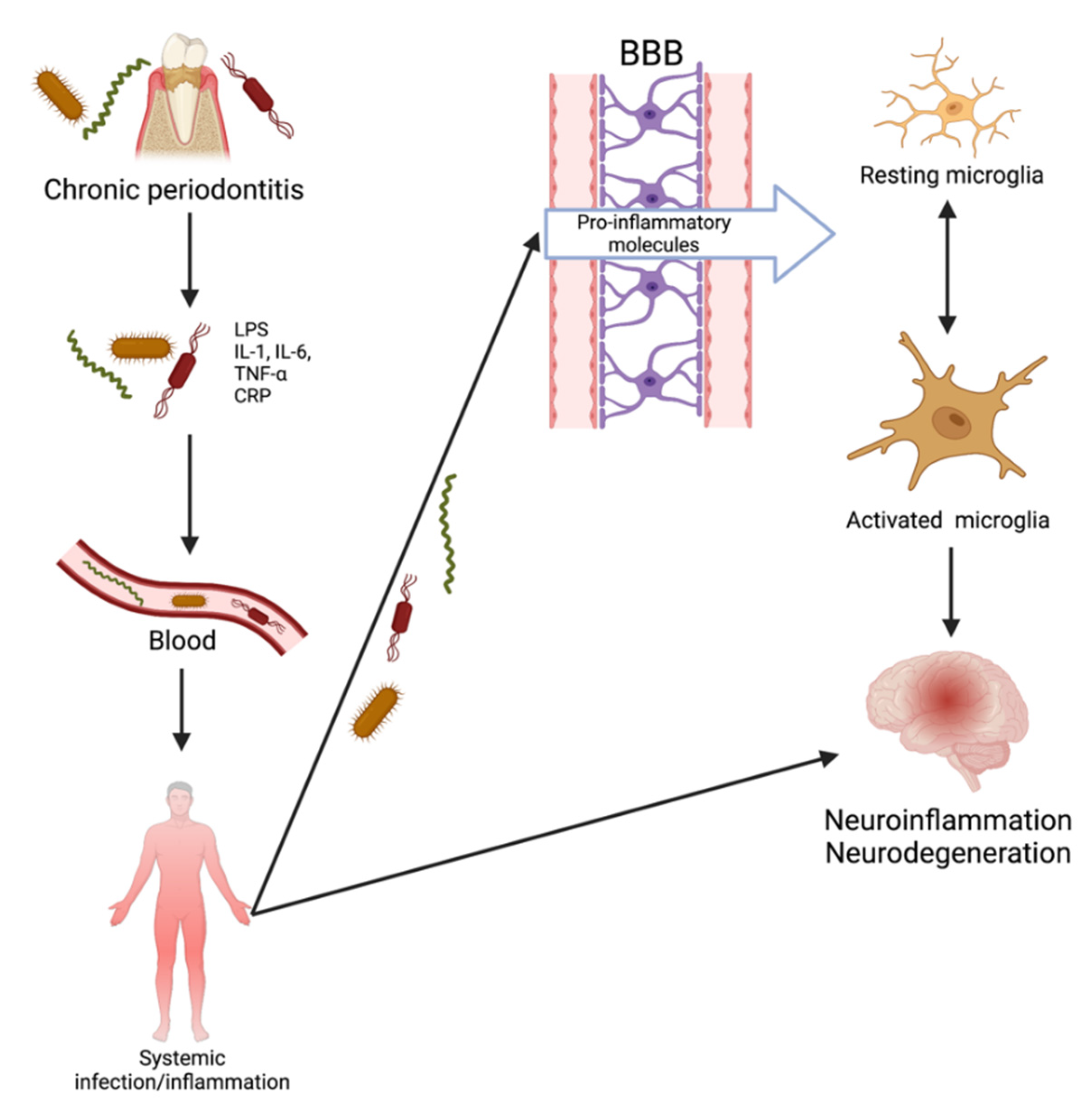
2.2. Oral Bacteria
2.3. Borrelia burgdorferi
2.4. Chlamydia pneumoniae
2.5. Fungal Pathogens
3. Conclusions
4. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.M.; Devenney, E.M.; Irish, M.; Ittner, A.; Naismith, S.; Ittner, L.; Rohrer, J.; Halliday, G.; Eisen, A.; Hodges, J.R.; et al. Neuronal network disintegration: Common pathways linking neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1234–1241. [Google Scholar] [CrossRef] [Green Version]
- Gary, C. Experimental Transmission of Alzheimer’s Disease Endophenotypes to Murine and Primate Models. Ph.D. Dissertation, Université Paris-Saclay, Gif-sur-Yvette, France, 2016. [Google Scholar]
- Prince, M.; Ali, G.C.; Guerchet, M.; Prina, A.M.; Albanese, E.; Wu, Y.T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers’ Res. Ther. 2016, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Powell, T. Health Policy and Dementia. Curr. Psychiatry Rep. 2018, 20, 4. [Google Scholar] [CrossRef]
- Wiley, J. Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar]
- Kochanek, K.D.; Xu, J.Q.; Arias, E. Mortality in the United States, 2019. In NCHS Data Brief, No 395; National Center for Health Statistics: Hyattsville, MD, USA, 2020. [Google Scholar]
- Dickerson, B.C.; McGinnis, S.M.; Xia, C.; Price, B.H.; Atri, A.; Murray, M.E.; Mendez, M.F.; Wolk, D.A. Approach to atypical Alzheimer’s disease and case studies of the major subtypes. CNS Spectr. 2017, 22, 439–449. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimers Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Montenegro, J.M.; Villarini, B.; Angelopoulou, A.; Kapetanios, E.; Garcia-Rodriguez, J.; Argyriou, V. A Survey of Alzheimer’s Disease Early Diagnosis Methods for Cognitive Assessment. Sensors 2020, 20, 7292. [Google Scholar] [CrossRef] [PubMed]
- Villain, N.; Dubois, B. Alzheimer’s Disease Including Focal Presentations. Semin. Neurol. 2019, 39, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Ljubenkov, P.A.; Geschwind, M.D. Dementia. Semin. Neurol. 2016, 36, 397–404. [Google Scholar] [PubMed] [Green Version]
- Shea, Y.F.; Pan, Y.; Mak, H.K.; Bao, Y.; Lee, S.C.; Chiu, P.K.; Chan, H.F. A systematic review of atypical Alzheimer’s disease including behavioural and psychological symptoms. Psychogeriatrics 2021, 21, 396–406. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; McHugh, P.F. FDG- and amyloid-PET in Alzheimer’s disease: Is the whole greater than the sum of the parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar]
- Bird, T.D. Alzheimer Disease Overview. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1998; [updated 2018]. [Google Scholar]
- Balin, B.J.; Hammond, C.J.; Little, C.S.; Hingley, S.T.; Al-Atrache, Z.; Appelt, D.M.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydia pneumoniae: An Etiologic Agent for Late-Onset Dementia. Front. Aging Neurosci. 2018, 10, 302. [Google Scholar] [CrossRef] [Green Version]
- Gomperts, S.N. Lewy Body Dementias: Dementia with Lewy Bodies and Parkinson Disease Dementia. Contin. Lifelong Learn. Neurol. 2016, 22, 435–463. [Google Scholar] [CrossRef]
- Sezgin, M.; Bilgic, B.; Tinaz, S.; Emre, M. Parkinson’s Disease Dementia and Lewy Body Disease. Semin. Neurol. 2019, 39, 274–282. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Rujeedawa, T.; Carrillo Felez, E.; Clare, I.C.H.; Fortea, J.; Strydom, A.; Rebillat, A.S.; Coppus, A.; Levin, J.; Zaman, S.H. The Clinical and Neuropathological Features of Sporadic (Late-Onset) and Genetic Forms of Alzheimer’s Disease. J. Clin. Med. 2021, 10, 4582. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Itzhaki, R.F.; Balin, B.J.; Miklossy, J.; Barron, A.E. Role of Microbes in the Development of Alzheimer’s Disease: State of the Art—An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front. Genet. 2018, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D. Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 591–601. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.; Fenech, M. A review of genome mutation and Alzheimer’s disease. Mutagenesis 2007, 22, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [Green Version]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; Del-Favero, J.; Cruts, M.; van Duijn, C.M.; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006, 129, 2977–2983. [Google Scholar] [CrossRef] [Green Version]
- Rumble, B.; Retallack, R.; Hilbich, C.; Simms, G.; Multhaup, G.; Martins, R.; Hockey, A.; Montgomery, P.; Beyreuther, K.; Masters, C.L. Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. N. Engl. J. Med. 1989, 320, 1446–1452. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; Dalton, A.J.; McLachlan, C.; Wen, G.Y.; Wisniewski, H.M. Alzheimer’s disease in Down’s syndrome: Clinicopathologic studies. Neurology 1985, 35, 957–961. [Google Scholar] [CrossRef] [Green Version]
- Alic, I.; Goh, P.A.; Murray, A.; Portelius, E.; Gkanatsiou, E.; Gough, G.; Mok, K.Y.; Koschut, D.; Brunmeir, R.; Yeap, Y.J.; et al. Patient-specific Alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose-sensitive AD suppressor in human brain. Mol. Psychiatry 2021, 26, 5766–5788. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. JINS 2017, 23, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front. Aging Neurosci. 2018, 10, 224. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; de Leon, M.J. Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol 2000 2020, 83, 242–271. [Google Scholar] [CrossRef] [PubMed]
- GA, H.J.H. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Kandimalla, R.J.; Prabhakar, S.; Wani, W.Y.; Kaushal, A.; Gupta, N.; Sharma, D.R.; Grover, V.K.; Bhardwaj, N.; Jain, K.; Gill, K.D. CSF p-Tau levels in the prediction of Alzheimer’s disease. Biol. Open 2013, 2, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Barichello, T.; Generoso, J.S.; Goularte, J.A.; Collodel, A.; Pitcher, M.R.; Simoes, L.R.; Quevedo, J.; Dal-Pizzol, F. Does Infection-Induced Immune Activation Contribute to Dementia? Aging Dis. 2015, 6, 342–348. [Google Scholar]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C.; et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 2009, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Priya, R.; Brutkiewicz, R.R. Brain astrocytes and microglia express functional MR1 molecules that present microbial antigens to mucosal-associated invariant T (MAIT) cells. J. Neuroimmunol. 2020, 349, 577428. [Google Scholar] [CrossRef]
- Dusseaux, M.; Martin, E.; Serriari, N.; Peguillet, I.; Premel, V.; Louis, D.; Milder, M.; Le Bourhis, L.; Soudais, C.; Treiner, E.; et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 2011, 117, 1250–1259. [Google Scholar] [CrossRef]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Huang, S.; Gilfillan, S.; Cella, M.; Miley, M.J.; Lantz, O.; Lybarger, L.; Fremont, D.H.; Hansen, T.H. Evidence for MR1 antigen presentation to mucosal-associated invariant T cells. J. Biol. Chem. 2005, 280, 21183–21193. [Google Scholar] [CrossRef] [Green Version]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Karamooz, E.; Harriff, M.J.; Lewinsohn, D.M. MR1-dependent antigen presentation. Semin. Cell Dev. Biol. 2018, 84, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Brutkiewicz, R.R. The Toll-like receptor 9 signalling pathway regulates MR1-mediated bacterial antigen presentation in B cells. Immunology 2017, 152, 232–242. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.E.; Villadangos, J.A. MR1 antigen presentation to MAIT cells: New ligands, diverse pathways? Curr. Opin. Immunol. 2018, 52, 108–113. [Google Scholar] [CrossRef]
- Ussher, J.E.; van Wilgenburg, B.; Hannaway, R.F.; Ruustal, K.; Phalora, P.; Kurioka, A.; Hansen, T.H.; Willberg, C.B.; Phillips, R.E.; Klenerman, P. TLR signaling in human antigen-presenting cells regulates MR1-dependent activation of MAIT cells. Eur. J. Immunol. 2016, 46, 1600–1614. [Google Scholar] [CrossRef] [Green Version]
- Kurioka, A.; Ussher, J.E.; Cosgrove, C.; Clough, C.; Fergusson, J.R.; Smith, K.; Kang, Y.H.; Walker, L.J.; Hansen, T.H.; Willberg, C.B.; et al. MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol. 2015, 8, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Le Bourhis, L.; Dusseaux, M.; Bohineust, A.; Bessoles, S.; Martin, E.; Premel, V.; Core, M.; Sleurs, D.; Serriari, N.E.; Treiner, E.; et al. MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog. 2013, 9, e1003681. [Google Scholar] [CrossRef] [Green Version]
- Leeansyah, E.; Svard, J.; Dias, J.; Buggert, M.; Nystrom, J.; Quigley, M.F.; Moll, M.; Sonnerborg, A.; Nowak, P.; Sandberg, J.K. Arming of MAIT Cell Cytolytic Antimicrobial Activity Is Induced by IL-7 and Defective in HIV-1 Infection. PLoS Pathog. 2015, 11, e1005072. [Google Scholar] [CrossRef]
- Held, K.; Bhonsle-Deeng, L.; Siewert, K.; Sato, W.; Beltran, E.; Schmidt, S.; Ruhl, G.; Ng, J.K.; Engerer, P.; Moser, M.; et al. Alphabeta T-cell receptors from multiple sclerosis brain lesions show MAIT cell-related features. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e107. [Google Scholar] [CrossRef] [Green Version]
- Willing, A.; Leach, O.A.; Ufer, F.; Attfield, K.E.; Steinbach, K.; Kursawe, N.; Piedavent, M.; Friese, M.A. CD8+ MAIT cells infiltrate into the CNS and alterations in their blood frequencies correlate with IL-18 serum levels in multiple sclerosis. Eur. J. Immunol. 2014, 44, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.L.; McPhie, D.L.; Chen, Y. Alzheimer’s disease: Dysfunction of a signalling pathway mediated by the amyloid precursor protein? Biochem. Soc. Symp. 2001, 67, 37–50. [Google Scholar]
- Miklossy, J. Alzheimer’s disease—A spriochetosis? Neuroreport 1993, 4, 841–848. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.B.; Miranda, J.M. Concurrent neocortical borreliosis and Alzheimer’s disease. Hum. Pathol. 1987, 18, 759–761. [Google Scholar] [CrossRef]
- Pisa, D.; Alonso, R.; Fernandez-Fernandez, A.M.; Rabano, A.; Carrasco, L. Polymicrobial Infections in Brain Tissue from Alzheimer’s Disease Patients. Sci. Rep. 2017, 7, 5559. [Google Scholar] [CrossRef]
- Calkosinski, I.; Dobrzynski, M.; Calkosinska, M.; Seweryn, E.; Bronowicka-Szydelko, A.; Dzierzba, K.; Ceremuga, I.; Gamian, A. [Characterization of an inflammatory response]. Postepy Hig. Med. Dosw. 2009, 63, 395–408. [Google Scholar]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Rutsch, A.; Kantsjo, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Harris, S.A.; Harris, E.A. Herpes Simplex Virus Type 1 and Other Pathogens are Key Causative Factors in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 319–353. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016; National Center for Health Statistics: Hyattsville, MD, USA, 2018; pp. 1–8.
- Suzich, J.B.; Cliffe, A.R. Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 2018, 522, 81–91. [Google Scholar] [CrossRef]
- Ball, M.J. Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved? Can. J. Neurol. Sci. 1982, 9, 303–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhou, Z.; Wang, H.; Cheng, X.; Zhong, S.; Zhao, C. Association of apolipoprotein E genotypes with epilepsy risk: A systematic review and meta-analysis. Epilepsy Behav. 2019, 98, 27–35. [Google Scholar] [CrossRef]
- Lamoureux, L.; Marottoli, F.M.; Tseng, K.Y.; Tai, L.M. APOE4 Promotes Tonic-Clonic Seizures, an Effect Modified by Familial Alzheimer’s Disease Mutations. Front. Cell Dev. Biol. 2021, 9, 656521. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Craske, J.; Wilcock, G.K.; Itzhaki, R.F. Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer’s disease brain using polymerase chain reaction. Biochem. Soc. Trans. 1991, 19, 122S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014, 6, 202. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious Agents and Neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, A.N.; Itzhaki, R.F.; Wozniak, M.A.; Patel, R.; Smit, E.J.; Noone, R.; Gilleran, G.; Taylor, S.; White, D.J. Apolipoprotein E-epsilon 4 and recurrent genital herpes in individuals co-infected with herpes simplex virus type 2 and HIV. Sex. Transm. Infect. 2008, 84, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Mancuso, R.; Baglio, F.; Cabinio, M.; Hernis, A.; Costa, A.S.; Calabrese, E.; Nemni, R.; Clerici, M. High avidity HSV-1 antibodies correlate with absence of amnestic Mild Cognitive Impairment conversion to Alzheimer’s disease. Brain Behav. Immun. 2016, 58, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Letenneur, L.; Peres, K.; Fleury, H.; Garrigue, I.; Barberger-Gateau, P.; Helmer, C.; Orgogozo, J.M.; Gauthier, S.; Dartigues, J.F. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: A population-based cohort study. PLoS ONE 2008, 3, e3637. [Google Scholar] [CrossRef] [Green Version]
- Lövheim, H.; Gilthorpe, J.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimer’s Dement. 2015, 11, 587–592. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 2017, 31, 3216–3226. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Carbone, I.; Raschi, E.; Porcellini, E. The 21st century epidemic: Infections as inductors of neuro-degeneration associated with Alzheimer’s Disease. Immun. Ageing 2014, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections—A Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e67. [Google Scholar] [CrossRef] [Green Version]
- Komaroff, A.L.; Boeckh, M.; Eliason, E.; Phan, T.; Kaufer, B.B. Summary of the 10th International Conference on Human Herpesviruses-6 and -7 (HHV-6A, -6B, and HHV-7). J. Med. Virol. 2018, 90, 625–630. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, L.; Zhu, Q.; Shu, M.; Cai, Q. Common Infections May Lead to Alzheimer’s Disease. Virol. Sin. 2018, 33, 456–458. [Google Scholar] [CrossRef]
- Fenno, J.C. Treponema denticola interactions with host proteins. J. Oral. Microbiol. 2012, 4, 9929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mira, A.; Simon-Soro, A.; Curtis, M.A. Role of microbial communities in the pathogenesis of periodontal diseases and caries. J. Clin. Periodontol. 2017, 44 (Suppl. 18), S23–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, A.R.; Dasanayake, A.P.; Craig, R.G.; Glodzik-Sobanska, L.; Bry, M.; de Leon, M.J. Alzheimer’s disease and peripheral infections: The possible contribution from periodontal infections, model and hypothesis. J. Alzheimer’s Dis. 2008, 13, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Abbayya, K.; Puthanakar, N.Y.; Naduwinmani, S.; Chidambar, Y.S. Association between Periodontitis and Alzheimer’s Disease. N. Am. J. Med. Sci. 2015, 7, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa-Ishii, S.; Shimada, A.; Imamura, F. Lipopolysaccharide-initiated persistent rhinitis causes gliosis and synaptic loss in the olfactory bulb. Sci. Rep. 2017, 7, 11605. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, N.S.; Chung, C.H.; Yeh, C.B.; Huang, R.Y.; Yuh, D.Y.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Chiang, W.S.; et al. Are Chronic Periodontitis and Gingivitis Associated with Dementia? A Nationwide, Retrospective, Matched-Cohort Study in Taiwan. Neuroepidemiology 2016, 47, 82–93. [Google Scholar] [CrossRef]
- Lee, Y.L.; Hu, H.Y.; Huang, L.Y.; Chou, P.; Chu, D. Periodontal Disease Associated with Higher Risk of Dementia: Population-Based Cohort Study in Taiwan. J. Am. Geriatr. Soc. 2017, 65, 1975–1980. [Google Scholar] [CrossRef]
- Naorungroj, S.; Schoenbach, V.J.; Wruck, L.; Mosley, T.H.; Gottesman, R.F.; Alonso, A.; Heiss, G.; Beck, J.; Slade, G.D. Tooth loss, periodontal disease, and cognitive decline in the Atherosclerosis Risk in Communities (ARIC) study. Community Dent. Oral Epidemiol. 2015, 43, 47–57. [Google Scholar] [CrossRef]
- Kaye, E.K.; Valencia, A.; Baba, N.; Spiro, A., 3rd; Dietrich, T.; Garcia, R.I. Tooth loss and periodontal disease predict poor cognitive function in older men. J. Am. Geriatr. Soc. 2010, 58, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.S.; Desrosiers, M.; Donegan, S.J.; Yepes, J.F.; Kryscio, R.J. Tooth loss, dementia and neuropathology in the Nun study. J. Am. Dent. Assoc. 2007, 138, 1314–1322, quiz 1381–1312. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Weyant, R.J.; Garcia, M.E.; Harris, T.; Launer, L.J.; Satterfield, S.; Simonsick, E.M.; Yaffe, K.; Newman, A.B. Adverse oral health and cognitive decline: The health, aging and body composition study. J. Am. Geriatr. Soc. 2013, 61, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, K.; Chang, J.; Kim, S.M.; Kim, S.J.; Cho, H.J.; Park, S.M. Association of Chronic Periodontitis on Alzheimer’s Disease or Vascular Dementia. J. Am. Geriatr. Soc. 2019, 67, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.T.; Lee, H.C.; Hu, C.J.; Huang, L.K.; Chao, S.P.; Lin, C.P.; Su, E.C.Y.; Lee, Y.C.; Chen, C.C. Periodontitis as a modifiable risk factor for dementia: A nationwide population-based cohort study. J. Am. Geriatr. Soc. 2017, 65, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Wu, Y.T.; Chang, Y.C. Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimer’s Res. Ther. 2017, 9, 56. [Google Scholar] [CrossRef]
- Arrive, E.; Letenneur, L.; Matharan, F.; Laporte, C.; Helmer, C.; Barberger-Gateau, P.; Miquel, J.L.; Dartigues, J.F. Oral health condition of French elderly and risk of dementia: A longitudinal cohort study. Community Dent. Oral Epidemiol. 2012, 40, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Yoshihara, A.; Kimura, Y.; Sato, M.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fukutomi, E.; Chen, W.; Imai, H.; et al. Longitudinal relationship of severe periodontitis with cognitive decline in older Japanese. J. Periodontal Res. 2016, 51, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Kimura, Y.; Ogawa, H.; Yamaga, T.; Ansai, T.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fujisawa, M.; Okumiya, K.; et al. Periodontitis, periodontal inflammation, and mild cognitive impairment: A 5-year cohort study. J. Periodontal Res. 2019, 54, 233–240. [Google Scholar] [CrossRef]
- Nilsson, H.; Sanmartin Berglund, J.; Renvert, S. Longitudinal evaluation of periodontitis and development of cognitive decline among older adults. J. Clin. Periodontol 2018, 45, 1142–1149. [Google Scholar] [CrossRef]
- Okamoto, N.; Morikawa, M.; Tomioka, K.; Yanagi, M.; Amano, N.; Kurumatani, N. Association between tooth loss and the development of mild memory impairment in the elderly: The Fujiwara-kyo Study. J. Alzheimer’s Dis. 2015, 44, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.S.; Shin, M.S.; Ahn, Y.B.; Choi, B.Y.; Nam, J.H.; Kim, H.D. Periodontitis is. associated with cognitive impairment in elderly Koreans: Results from the Yangpyeong Cohort Study. J. Am. Geriatr. Soc. 2016, 64, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Morikawa, M.; Amano, N.; Yanagi, M.; Takasawa, S.; Kurumatani, N. Effects of Tooth Loss and the Apolipoprotein E varepsilon4 Allele on Mild Memory Impairment in the Fujiwara-kyo Study of Japan: A Nested Case-Control Study. J. Alzheimer’s Dis. 2017, 55, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Leira, Y.; Iglesias-Rey, R.; Gomez-Lado, N.; Aguiar, P.; Campos, F.; D’Aiuto, F.; Castillo, J.; Blanco, J.; Sobrino, T. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch. Oral Biol. 2019, 99, 120–125. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Ren, J.; Yu, H.; Yu, W.; Zhou, Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun. Ageing 2018, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflamm. 2018, 15, 37. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Chukkapalli, S.; Poole, S.; Velsko, I.; Crean, S.J.; Kesavalu, L. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE(−)(/)(−) mice brains. J. Oral Microbiol. 2017, 9, 1270602. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Z.; Nakanishi, Y.; Ni, J.; Hayashi, Y.; Takayama, F.; Zhou, Y.; Kadowaki, T.; Nakanishi, H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017, 7, 11759. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J. Alzheimer’s Dis. 2015, 43, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklossy, J. Bacterial Amyloid and DNA are Important Constituents of Senile Plaques: Further Evidence of the Spirochetal and Biofilm Nature of Senile Plaques. J. Alzheimer’s Dis. 2016, 53, 1459–1473. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Pacheco e Silva, A.C. Espirochetose dos centros nervos. Mem. Hospicio Juquery 1926, 3–4, 1–27. [Google Scholar]
- Kugeler, K.J.; Schwartz, A.M.; Delorey, M.J.; Mead, P.S.; Hinckley, A.F. Estimating the Frequency of Lyme Disease Diagnoses, United States, 2010–2018. Emerg. Infect. Dis. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Schwartz, A.M.; Kugeler, K.J.; Nelson, C.A.; Marx, G.E.; Hinckley, A.F. Use of Commercial Claims Data for Evaluating Trends in Lyme Disease Diagnoses, United States, 2010–2018. Emerg. Infect. Dis. 2021, 27, 499–507. [Google Scholar] [CrossRef]
- Ramesh, G.; Borda, J.T.; Dufour, J.; Kaushal, D.; Ramamoorthy, R.; Lackner, A.A.; Philipp, M.T. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. Am. J. Pathol. 2008, 173, 1415–1427. [Google Scholar] [CrossRef] [Green Version]
- Luft, B.J.; Steinman, C.R.; Neimark, H.C.; Muralidhar, B.; Rush, T.; Finkel, M.F.; Kunkel, M.; Dattwyler, R.J. Invasion of the central nervous system by Borrelia burgdorferi in acute disseminated infection. JAMA 1992, 267, 1364–1367. [Google Scholar] [CrossRef]
- Maksimyan, S.; Syed, M.S.; Soti, V. Post-Treatment Lyme Disease Syndrome: Need for Diagnosis and Treatment. Cureus 2021, 13, e18703. [Google Scholar] [CrossRef] [PubMed]
- Crossland, N.A.; Alvarez, X.; Embers, M.E. Late Disseminated Lyme Disease: Associated Pathology and Spirochete Persistence Posttreatment in Rhesus Macaques. Am. J. Pathol. 2018, 188, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Embers, M.E.; Barthold, S.W.; Borda, J.T.; Bowers, L.; Doyle, L.; Hodzic, E.; Jacobs, M.B.; Hasenkampf, N.R.; Martin, D.S.; Narasimhan, S.; et al. Persistence of Borrelia burgdorferi in rhesus macaques following antibiotic treatment of disseminated infection. PLoS ONE 2012, 7, e29914. [Google Scholar] [CrossRef]
- Hodzic, E.; Imai, D.; Feng, S.; Barthold, S.W. Resurgence of persisting non-cultivable Borrelia burgdorferi following antibiotic treatment in mice. PLoS ONE 2014, 9, e86907. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Monco, J.C.; Villar, B.F.; Alen, J.C.; Benach, J.L. Borrelia burgdorferi in the central nervous system: Experimental and clinical evidence for early invasion. J. Infect. Dis. 1990, 161, 1187–1193. [Google Scholar] [CrossRef]
- Meer-Scherrer, L.; Chang Loa, C.; Adelson, M.E.; Mordechai, E.; Lobrinus, J.A.; Fallon, B.A.; Tilton, R.C. Lyme disease associated with Alzheimer’s disease. Curr. Microbiol. 2006, 52, 330–332. [Google Scholar] [CrossRef]
- Miklossy, J.; Khalili, K.; Gern, L.; Ericson, R.L.; Darekar, P.; Bolle, L.; Hurlimann, J.; Paster, B.J. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J. Alzheimer’s Dis. 2004, 6, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J. Alzheimer’s disease—A neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflamm. 2011, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Gadila, S.K.G.; Rosoklija, G.; Dwork, A.J.; Fallon, B.A.; Embers, M.E. Detecting Borrelia Spirochetes: A Case Study with Validation among Autopsy Specimens. Front. Neurol. 2021, 12, 628045. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Omar, R.; Saran, B.; Andorn, A.; Suarez, M.; Pavia, C.; Weinstein, A.; Shank, D.; Davis, K.; Burgdorfer, W. Concurrent neuroborreliosis and Alzheimer’s disease: Analysis of the evidence. Hum. Pathol. 1989, 20, 753–757. [Google Scholar] [CrossRef]
- Gutacker, M.; Valsangiacomo, C.; Balmelli, T.; Bernasconi, M.V.; Bouras, C.; Piffaretti, J.C. Arguments against the involvement of Borrelia burgdorferi sensu lato in Alzheimer’s disease. Res. Microbiol. 1998, 149, 31–37. [Google Scholar] [CrossRef]
- Marques, A.R.; Weir, S.C.; Fahle, G.A.; Fischer, S.H. Lack of evidence of Borrelia involvement in Alzheimer’s disease. J. Infect. Dis. 2000, 182, 1006–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, R.; Kin, N.M.; Chen, M.F.; Nair, N.P.; Chan, E.C. Alzheimer’s disease may not be a spirochetosis. Neuroreport 1999, 1, 1489–1491. [Google Scholar] [CrossRef]
- Galbussera, A.; Tremolizzo, L.; Isella, V.; Gelosa, G.; Vezzo, R.; Vigorè, L.; Brenna, M.; Ferrarese, C.; Appollonio, I. Lack of evidence for Borrelia burgdorferi seropositivity in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 308. [Google Scholar] [CrossRef] [PubMed]
- Kugeler, K.J.; Farley, G.M.; Forrester, J.D.; Mead, P.S. Geographic Distribution and Expansion of Human Lyme Disease, United States. Emerg. Infect. Dis. 2015, 21, 1455–1457. [Google Scholar] [CrossRef]
- Schumacher, H.R., Jr.; Gerard, H.C.; Arayssi, T.K.; Pando, J.A.; Branigan, P.J.; Saaibi, D.L.; Hudson, A.P. Lower prevalence of Chlamydia pneumoniae DNA compared with Chlamydia trachomatis DNA in synovial tissue of arthritis patients. Arthritis Rheum. 1999, 42, 1889–1893. [Google Scholar] [CrossRef]
- Belland, R.J.; Ouellette, S.P.; Gieffers, J.; Byrne, G.I. Chlamydia pneumoniae and atherosclerosis. Cell Microbiol. 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Wagner, A.D.; Gerard, H.C.; Fresemann, T.; Schmidt, W.A.; Gromnica-Ihle, E.; Hudson, A.P.; Zeidler, H. Detection of Chlamydia pneumoniae in giant cell vasculitis and correlation with the topographic arrangement of tissue-infiltrating dendritic cells. Arthritis Rheum. 2000, 43, 1543–1551. [Google Scholar] [CrossRef]
- Shima, K.; Kuhlenbaumer, G.; Rupp, J. Chlamydia pneumoniae infection and Alzheimer’s disease: A connection to remember? Med. Microbiol. Immunol. 2010, 199, 283–289. [Google Scholar] [CrossRef]
- Little, C.S.; Joyce, T.A.; Hammond, C.J.; Matta, H.; Cahn, D.; Appelt, D.M.; Balin, B.J. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 2014, 6, 304. [Google Scholar] [CrossRef] [Green Version]
- Balin, B.J.; Gérard, H.C.; Arking, E.J.; Appelt, D.M.; Branigan, P.J.; Abrams, J.T.; Whittum-Hudson, J.A.; Hudson, A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 1998, 187, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Gerard, H.C.; Dreses-Werringloer, U.; Wildt, K.S.; Deka, S.; Oszust, C.; Balin, B.J.; Frey, W.H., 2nd; Bordayo, E.Z.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 2006, 48, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelt, D.M.; Roupas, M.R.; Way, D.S.; Bell, M.G.; Albert, E.V.; Hammond, C.J.; Balin, B.J. Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci. 2008, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, C.J.; Hallock, L.R.; Howanski, R.J.; Appelt, D.M.; Little, C.S.; Balin, B.J. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 2010, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Boelen, E.; Steinbusch, H.W.; van der Ven, A.J.; Grauls, G.; Bruggeman, C.A.; Stassen, F.R. Chlamydia pneumoniae infection of brain cells: An in vitro study. Neurobiol. Aging 2007, 28, 524–532. [Google Scholar] [CrossRef]
- MacIntyre, A.; Hammond, C.J.; Little, C.S.; Appelt, D.M.; Balin, B.J. Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiol. Lett. 2002, 217, 167–172. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, A.; Abramov, R.; Hammond, C.J.; Hudson, A.P.; Arking, E.J.; Little, C.S.; Appelt, D.M.; Balin, B.J. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J. Neurosci. Res. 2003, 71, 740–750. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Gerard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae infection of human astrocytes and microglia in culture displays an active, rather than a persistent, phenotype. Am. J. Med. Sci. 2006, 332, 168–174. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Bhuiyan, M.; Zhao, Y.; Gerard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P. Initial characterization of Chlamydophila (Chlamydia) pneumoniae cultured from the late-onset Alzheimer brain. Int. J. Med. Microbiol. 2009, 299, 187–201. [Google Scholar] [CrossRef] [Green Version]
- Arking, E.J.; Appelt, D.M.; Abrams, J.T.; Kolbe, S.; Hudson, A.P.; Balin, B.J. Ultrastructural Analysis of Chlamydia pneumoniae in the Alzheimer’s Brain. Pathogenesis 1999, 1, 201–211. [Google Scholar]
- Paradowski, B.; Jaremko, M.; Dobosz, T.; Leszek, J.; Noga, L. Evaluation of CSF-Chlamydia pneumoniae, CSF-tau, and CSF-Abeta42 in Alzheimer’s disease and vascular dementia. J. Neurol. 2007, 254, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Nochlin, D.; Shaw, C.M.; Campbell, L.A.; Kuo, C.C. Failure to detect Chlamydia pneumoniae in brain tissues of Alzheimer’s disease. Neurology 1999, 53, 1888. [Google Scholar] [CrossRef]
- Gieffers, J.; Reusche, E.; Solbach, W.; Maass, M. Failure to detect Chlamydia pneumoniae in brain sections of Alzheimer’s disease patients. J. Clin. Microbiol. 2000, 38, 881–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsay, S.; Williams, S.; Mu, Y.; Epson, E.; Johnston, H.; Farley, M.M.; Harrison, L.H.; Vonbank, B.; Shrum, S.; Dumyati, G.; et al. National Burden of Candidemia, United States, 2017. Open Forum Infect. Dis. 2018, 5, S142–S143. [Google Scholar] [CrossRef] [Green Version]
- Parady, B. Innate Immune and Fungal Model of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2018, 2, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Pisa, D.; Alonso, R.; Rabano, A.; Rodal, I.; Carrasco, L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef] [Green Version]
- Pisa, D.; Alonso, R.; Rabano, A.; Carrasco, L. Corpora Amylacea of Brain Tissue from Neurodegenerative Diseases Are Stained with Specific Antifungal Antibodies. Front. Neurosci. 2016, 10, 86. [Google Scholar] [CrossRef] [Green Version]
- Pisa, D.; Alonso, R.; Juarranz, A.; Rabano, A.; Carrasco, L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Pisa, D.; Rabano, A.; Carrasco, L. Alzheimer’s disease and disseminated mycoses. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1125–1132. [Google Scholar] [CrossRef]
- Pisa, D.; Alonso, R.; Rabano, A.; Horst, M.N.; Carrasco, L. Fungal Enolase, beta-Tubulin, and Chitin Are Detected in Brain Tissue from Alzheimer’s Disease Patients. Front. Microbiol. 2016, 7, 1772. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.O.; Christianson, T.J.; Kremers, W.K.; Mielke, M.M.; Machulda, M.M.; Vassilaki, M.; Alhurani, R.E.; Geda, Y.E.; Knopman, D.S.; Petersen, R.C. Association Between Olfactory Dysfunction and Amnestic Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol. 2016, 73, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Chavez, M.J.; Perez-Garcia, L.A.; Nino-Vega, G.A.; Mora-Montes, H.M. Fungal Strategies to Evade the Host Immune Recognition. J. Fungi 2017, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Pisa, D.; Marina, A.I.; Morato, E.; Rabano, A.; Carrasco, L. Fungal infection in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 301–311. [Google Scholar] [CrossRef]
- Alonso, R.; Pisa, D.; Fernandez-Fernandez, A.M.; Carrasco, L. Infection of Fungi and Bacteria in Brain Tissue from Elderly Persons and Patients with Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 159. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landry, R.L.; Embers, M.E. Does Dementia Have a Microbial Cause? NeuroSci 2022, 3, 262-283. https://doi.org/10.3390/neurosci3020019
Landry RL, Embers ME. Does Dementia Have a Microbial Cause? NeuroSci. 2022; 3(2):262-283. https://doi.org/10.3390/neurosci3020019
Chicago/Turabian StyleLandry, Remi L., and Monica E. Embers. 2022. "Does Dementia Have a Microbial Cause?" NeuroSci 3, no. 2: 262-283. https://doi.org/10.3390/neurosci3020019
APA StyleLandry, R. L., & Embers, M. E. (2022). Does Dementia Have a Microbial Cause? NeuroSci, 3(2), 262-283. https://doi.org/10.3390/neurosci3020019