
Peptide-Functionalized Nanoparticles for the Targeted Delivery of Cytotoxins to MMP-14-Expressing Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. MMP-14 PEX Protein Purification
2.3. Preparation of Ansamitocin P-3 Encapsulated Cy3-HGC Nanoparticles
2.4. Physicochemical Characterization of Nanoparticles
2.5. Cell Culture and Transfection
2.6. Dot-Based Cell Migration Assay
2.7. MMP-14 Association and Endocytosis Mechanism Studies
2.8. Encapsulation Efficiency and Loading Capacity
2.9. Release Kinetics
2.10. Chicken Chorioallantoic Membrane (CAM) Assay
2.11. Statistical Analysis
3. Results
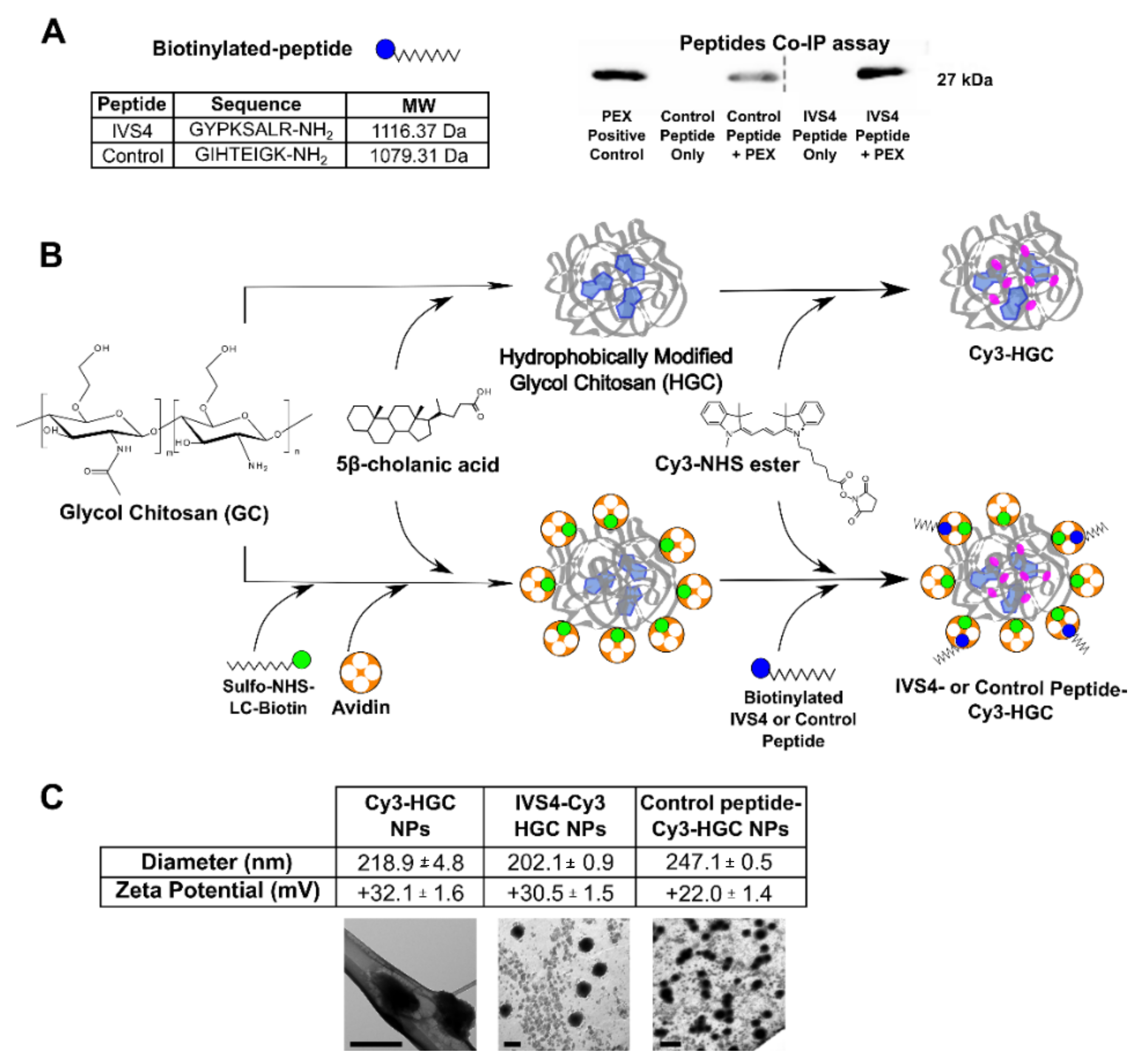
3.1. Development of MMP-14 Homing Peptide–Nanoparticle Delivery System
3.2. IVS4 Peptide Confers Selectivity to MMP-14-Expressing Cells
3.3. IVS4-Cy3-HGC NPs Bind to MMP-14 and Are Internalized via Endocytosis
3.4. Encapsulation of Cytotoxin Ansamitocin P-3 in IVS4-Cy3-HGC-NPs and System Characterization
3.5. AP3-Loaded IVS4-Cy3-HGC NPs Selectively Induce Cell Death in MMP-14-Expressing Cancer Cells
3.6. AP3-Loaded IVS4-Cy3-HGC NPs Prevent MMP-14-Expressing Cancer Cell Invasion In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinase |
| AP3 | ansamitocin P-3 |
| CAM | chorioallantoic membrane |
| DLS | dynamic light scattering |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | dimethylsulfoxide |
| DOX | doxorubicin |
| EDC | N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride |
| EEA-1 | early endosome antigen-1 |
| ECM | extracellular matrix |
| GC | glycol chitosan |
| HGC | hydrophobically modified glycol chitosan |
| HPLC | High-performance liquid chromatography |
| HRP | Horseradish peroxidase |
| LAMP-1 | lysosomal-associated membrane protein-1 |
| MMP-14 | matrix metalloproteinase-14 |
| MMPI | matrix metalloproteinase inhibitor |
| MT1-MMP | membrane type 1 matrix metalloproteinase |
| MWCO | molecular weight cut-off |
| NHS | N-hydroxysuccinimide |
| NP | nanoparticle |
| PBS | Phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PEX | hemopexin |
| SDS-PAGE | sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SIM | structure illumination microscopy |
| TEM | transmission electron microscopy |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of Matrix Metalloproteinase 14 (MMP-14)-mediated Cancer Cell Migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Chiarelli, C.; Kozarekar, P.; Adler, H.L. Membrane type 1-matrix metalloproteinase promotes human prostate cancer invasion and metastasis. Thromb. Haemost. 2005, 93, 770–778. [Google Scholar] [PubMed]
- Itoh, T.; Tanioka, M.; Yoshida, H.; Yoshioka, T.; Nishimoto, H.; Itohara, S. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998, 58, 1048–1051. [Google Scholar] [PubMed]
- Chun, T.-H.; Sabeh, F.; Ota, I.; Murphy, H.; McDonagh, K.T.; Holmbeck, K.; Birkedal-Hansen, H.; Allen, E.D.; Weiss, S.J. MT1-MMP–dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol. 2004, 167, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Tsunezuka, Y.; Kinoh, H.; Takino, T.; Watanabe, Y.; Okada, Y.; Shinagawa, A.; Sato, H.; Seiki, M. Expression of membrane-type matrix metalloproteinase 1 (MT1-MMP) in tumor cells enhances pulmonary metastasis in an experimental metastasis assay. Cancer Res. 1996, 56, 5678–5683. [Google Scholar] [PubMed]
- Yonemura, Y.; Endo, Y.; Takino, T.; Sakamoto, K.; Bandou, E.; Kinoshita, K.; Fushida, S.; Miwa, K.; Sugiyama, K.; Sasaki, T. Membrane-type 1 matrix metalloproteinase enhances lymph node metastasis of gastric cancer. Clin. Exp. Metastasis 2000, 18, 321–327. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Massova, I.; Kotra, L.P.; Fridman, R.; Mobashery, S. Matrix metalloproteinases: Structures, evolution, and diversification. FASEB J. 1998, 12, 1075–1095. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B.W. Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 1988, 21, 333–340. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Tassone, P.; Gozzini, A.; Goldmacher, V.; Shammas, M.A.; Whiteman, K.R.; Carrasco, D.R.; Li, C.; Allam, C.K.; Venuta, S.; Anderson, K.C. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 2004, 64, 4629–4636. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef] [Green Version]
- Venghateri, J.B.; Gupta, T.K.; Verma, P.J.; Kunwar, A.; Panda, D. Ansamitocin P3 depolymerizes microtubules and induces apoptosis by binding to tubulin at the vinblastine site. PLoS ONE 2013, 8, e75182. [Google Scholar]
- Ericsson, U.B.; Hallberg, B.M.; DeTitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006, 357, 289–298. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Hymowitz, M.; Conner, C.; Bahou, W.F.; Zucker, S. The propeptide domain of membrane type 1-matrix metalloproteinase acts as an intramolecular chaperone when expressed in trans with the mature sequence in COS-l cells. J. Biol. Chem. 2000, 275, 29648–29653. [Google Scholar] [CrossRef] [Green Version]
- Evensen, N.A.; Li, J.; Yang, J.; Yu, X.; Sampson, N.S.; Zucker, S.; Cao, J. Development of a high-throughput three-dimensional invasion assay for anti-cancer drug discovery. PLoS ONE 2013, 8, e8281. [Google Scholar] [CrossRef] [Green Version]
- Lokman, N.A.; Elder, A.S.; Ricciardelli, C.; Oehler, M.K. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef] [Green Version]
- Suarato, G.; Lee, S.I.; Li, W.; Rao, S.; Khan, T.; Meng, Y.; Shelly, M. Micellar nanocomplexes for biomagnetic delivery of intracellular proteins to dictate axon formation during neuronal development. Biomaterials 2017, 112, 176–191. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Li, W.; Rubenstein, D.A.; Meng, Y. Biocompatible and target specific hydrophobically modified glycol chitosan nanoparticles. Biointerphases 2016, 11, 04B301. [Google Scholar] [CrossRef]
- Jayakumar, R.; Chennazhi, K.P.; Muzzarelli, R.A.A.; Tamura, H.; Nair, S.V.; Selvamurugan, N. Chitosan conjugated DNA nanoparticles in gene therapy. Carbohydr. Polym. 2010, 79, 1–8. [Google Scholar] [CrossRef]
- Park, K.; Kim, J.-H.; Nam, Y.S.; Lee, S.; Nam, H.Y.; Kim, K.; Park, J.H.; Kim, I.-S.; Choi, K.; Kim, S.Y. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. J. Control. Release 2007, 122, 305–314. [Google Scholar] [CrossRef]
- Chin, A.; Suarato, G.; Meng, Y. Evaluation of physicochemical characteristics of hydrophobically modified glycol chitosan nano-particles and their biocompatibility in murine osteosarcoma and osteoblast-like cells. J. Nanotech. Smart Mater. 2014, 1, 1–7. [Google Scholar]
- Duceppe, N.; Tabrizian, M. Advances in using chitosan-based nanoparticles for in vitro and in vivo drug and gene delivery. Expert Opin. Drug Deliv. 2010, 7, 1191–1207. [Google Scholar] [CrossRef]
- Bacac, M.; Fusco, C.; Planche, A.; Santodomingo, J.; Demaurex, N.; Leemann-Zakaryan, R.; Provero, P.; Stamenkovic, I. Securin and separase modulate membrane traffic by affecting endosomal acidification. Traffic 2011, 12, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Salvati, A.; Åberg, C.; dos Santos, T.; Varela, J.; Pinto, P.; Lynch, I.; Dawson, K.A. Experimental and theoretical comparison of intracellular import of polymeric nanoparticles and small molecules: Toward models of uptake kinetics. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Nam, H.Y.; Kwon, S.M.; Chung, H.; Lee, S.-Y.; Kwon, S.-H.; Jeon, H.; Kim, Y.; Park, J.H.; Kim, J.; Her, S. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J. Control. Release 2009, 135, 259–267. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.J.; Chung, H.; Her, S.; Choi, Y.; Kim, K.; Choi, K.; Kwon, I.C. Cellular uptake pathway and drug release characteristics of drug-encapsulated glycol chitosan nanoparticles in live cells. Microsc. Res. Tech. 2010, 73, 857–865. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Wang, X.; Ma, D.; Keski-Oja, J.; Pei, D. Co-recycling of MT1-MMP and MT3-MMP through the Trans-Golgi Network: Identification of DKV582 as a recycling signal. J. Biol. Chem. 2004, 279, 9331–9336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacle, A.; Murphy, G.; Roghi, C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J. Cell Sci. 2003, 116, 3905–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Guo, Y.; Li, R.; Wang, T.; Han, M.; Zhu, C.; Wang, X. Methotrexate Nanoparticles Prepared with Codendrimer from Polyamidoamine (PAMAM) and Oligoethylene Glycols (OEG) Dendrons: Antitumor Efficacy In Vitro and In Vivo. Sci. Rep. 2016, 6, 28983. [Google Scholar] [CrossRef] [PubMed]
- Hrubý, M.; Koňák, Č.; Ulbrich, K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J. Control. Release 2005, 103, 137–148. [Google Scholar] [CrossRef]
- Missirlis, D.; Kawamura, R.; Tirelli, N.; Hubbell, J.A. Doxorubicin encapsulation and diffusional release from stable, polymeric, hydrogel nanoparticles. Eur. J. Pharm. Sci. 2006, 29, 120–129. [Google Scholar] [CrossRef]
- Wang, J.J.; Zeng, Z.W.; Xiao, R.Z.; Xie, T.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar]
- Min, K.H.; Park, K.; Kim, Y.-S.; Bae, S.M.; Lee, S.; Jo, H.G.; Park, R.-W.; Kim, I.-S.; Jeong, S.Y.; Kim, K.; et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J. Control. Release 2008, 127, 208–218. [Google Scholar] [CrossRef]
- Wolpert-Defilippes, M.; Adamson, R.; Cysyk, R.; Johns, D. Initial studies on the cytotoxic action of maytansine, a novel ansa macrolide. Biochem. Pharmacol. 1975, 24, 751–754. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Komoda, Y.; Court, W.; Thomas, G.; Smith, R.; Karim, A.; Gilmore, C.; Haltiwanger, R.; Bryan, R. Tumor inhibitors. LXXIII. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef]
- Li, J.; Zucker, S.; Pulkoski-Gross, A.; Kuscu, C.; Karaayvaz, M.; Ju, J.; Yao, H.; Song, E.; Cao, J. Conversion of stationary to invasive tumor initiating cells (TICs): Role of hypoxia in membrane type 1-matrix metalloproteinase (MT1-MMP) trafficking. PLoS ONE 2012, 7, e38403. [Google Scholar] [CrossRef] [Green Version]
- Nowak-Sliwinska, P.; Segura, T.; Iruela-Arispe, M.L. The chicken chorioallantoic membrane model in biology, medicine and bioengineering. Angiogenesis 2014, 17, 779–804. [Google Scholar] [CrossRef] [Green Version]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Scanlon, C.S.; Banerjee, R.; Russo, N.; Inglehart, R.C.; Willis, A.L.; Weiss, S.J.; D’Silva, N.J. The Histone Methyltransferase EZH2 Mediates Tumor Progression on the Chick Chorioallantoic Membrane Assay, a Novel Model of Head and Neck Squamous Cell Carcinoma. Transl. Oncol. 2013, 6, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Rowe, R.G.; Li, X.Y.; Hu, Y.; Saunders, T.L.; Virtanen, I.; de Herreros, A.G.; Becker, K.F.; Ingvarsen, S.; Engelholm, L.H.; Bommer, G.T.; et al. Mesenchymal cells reactivate Snail1 expression to drive three-dimensional invasion programs. J. Cell Biol. 2009, 184, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Helbig, G.; Christopherson, K.W.; Bhat-Nakshatri, P.; Kumar, S.; Kishimoto, H.; Miller, K.D.; Broxmeyer, H.E.; Nakshatri, H. NF-κ B promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J. Biol. Chem. 2003, 278, 21631–21638. [Google Scholar] [CrossRef] [Green Version]
- Suarato, G.; Li, W.; Meng, Y. Role of pH-responsiveness in the design of chitosan-based cancer nanotherapeutics: A review. Biointerphases 2016, 11, 04B201. [Google Scholar] [CrossRef]
- Li, W.Y.; Suarato, G.; Cathcart, J.M.; Sargunas, P.R.; Meng, Y.Z. Design, characterization, and intracellular trafficking of biofunctionalized chitosan nanomicelles. Biointerphases 2020, 15, 14. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cathcart, J.; Suarato, G.; Li, W.; Cao, J.; Meng, Y. Peptide-Functionalized Nanoparticles for the Targeted Delivery of Cytotoxins to MMP-14-Expressing Cancer Cells. Biophysica 2022, 2, 203-220. https://doi.org/10.3390/biophysica2030021
Cathcart J, Suarato G, Li W, Cao J, Meng Y. Peptide-Functionalized Nanoparticles for the Targeted Delivery of Cytotoxins to MMP-14-Expressing Cancer Cells. Biophysica. 2022; 2(3):203-220. https://doi.org/10.3390/biophysica2030021
Chicago/Turabian StyleCathcart, Jillian, Giulia Suarato, Weiyi Li, Jian Cao, and Yizhi Meng. 2022. "Peptide-Functionalized Nanoparticles for the Targeted Delivery of Cytotoxins to MMP-14-Expressing Cancer Cells" Biophysica 2, no. 3: 203-220. https://doi.org/10.3390/biophysica2030021
APA StyleCathcart, J., Suarato, G., Li, W., Cao, J., & Meng, Y. (2022). Peptide-Functionalized Nanoparticles for the Targeted Delivery of Cytotoxins to MMP-14-Expressing Cancer Cells. Biophysica, 2(3), 203-220. https://doi.org/10.3390/biophysica2030021