


Comparison of Empirical Zn2+ Models in Protein–DNA Complexes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Setup and Zn Models
2.1.1. 12-6-4 Lennard–Jones-Type Potential (12-6-4)
2.1.2. Cationic Dummy Atom Model (CDAM)
2.1.3. Extended Zinc AMBER Force Field (EZAFF)
2.2. Molecular Dynamics Simulations
2.3. Analysis
3. Results
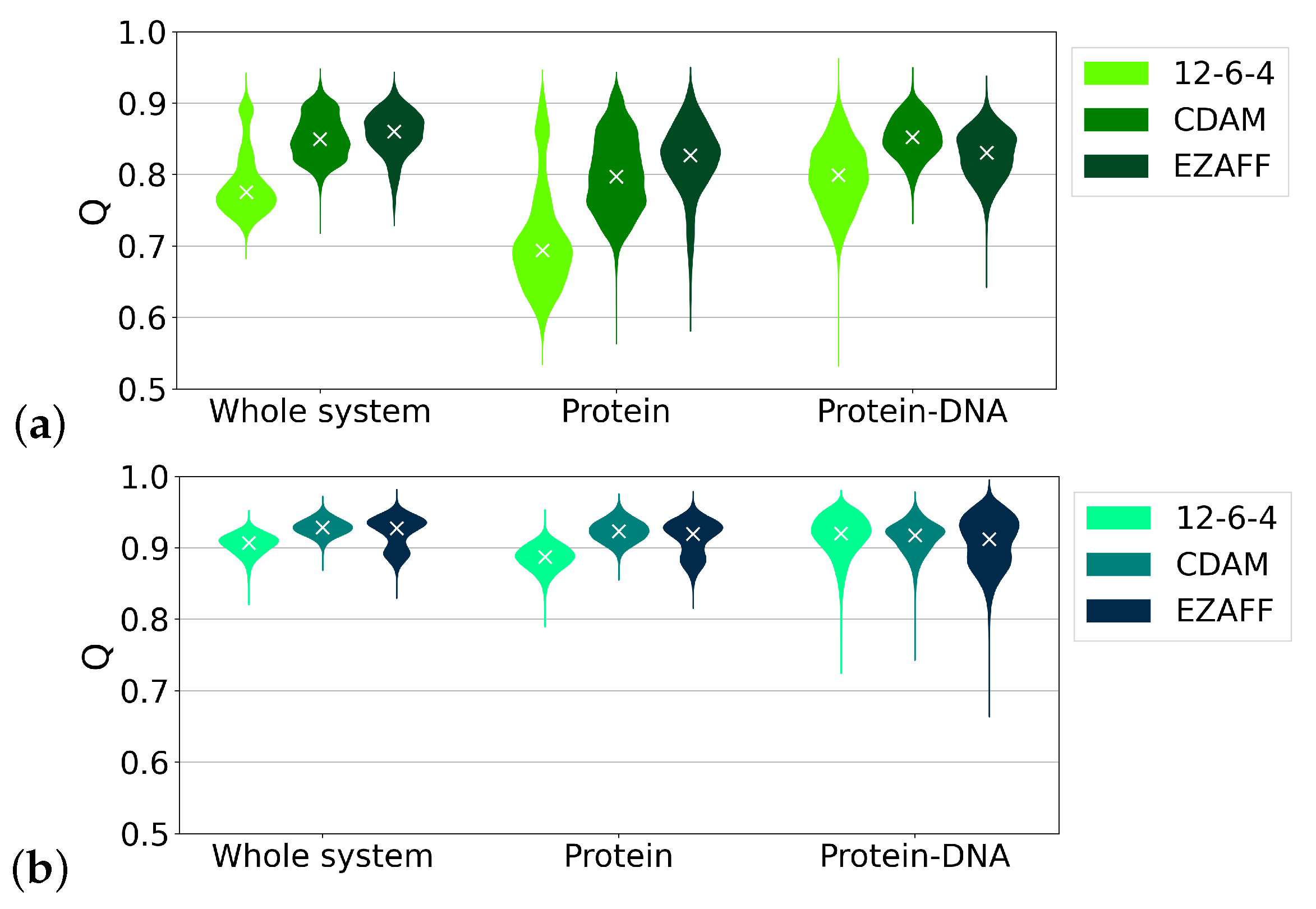
3.1. Protein Structure
3.2. DNA Structure
3.3. Protein–DNA Interactions
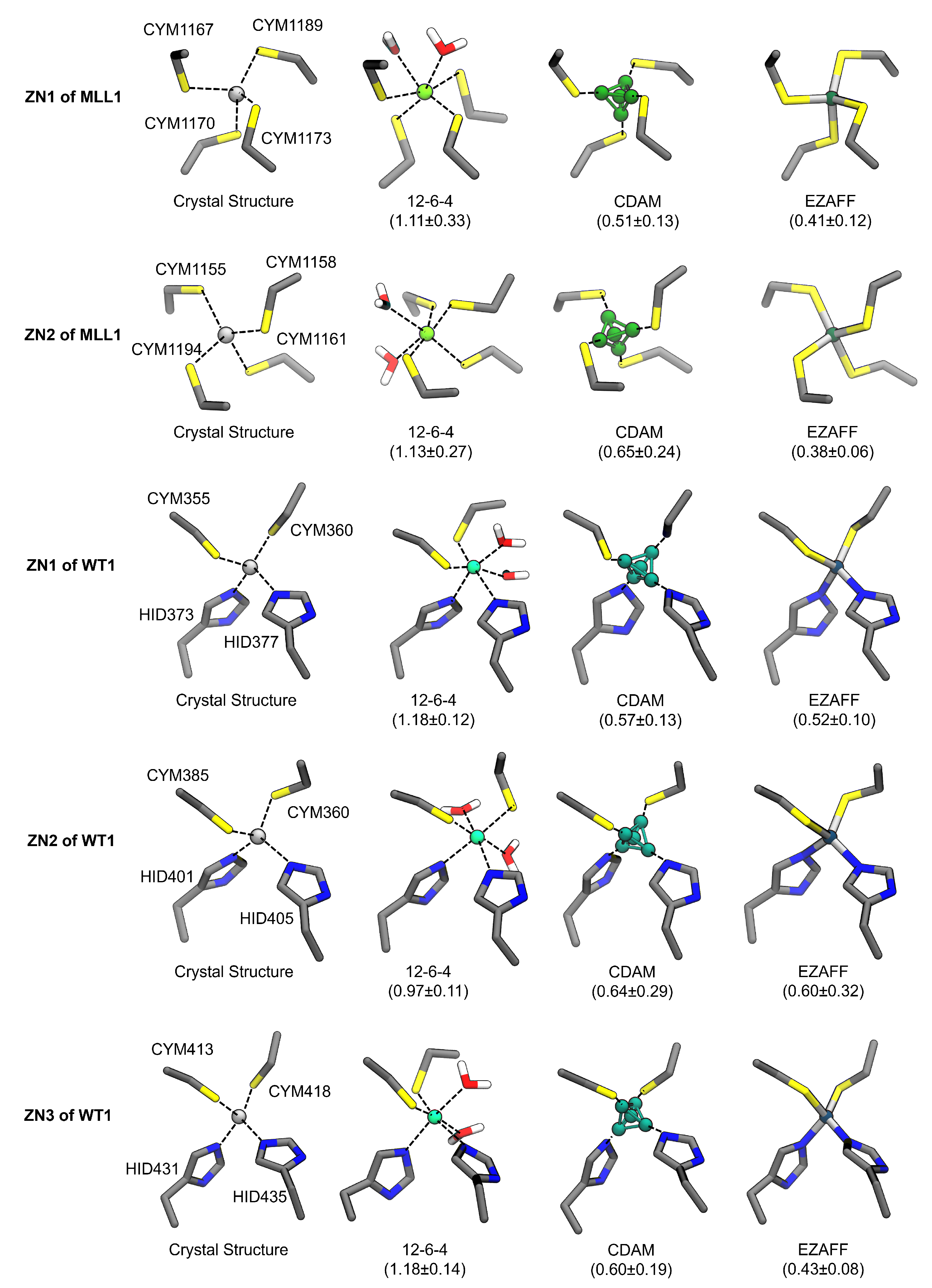
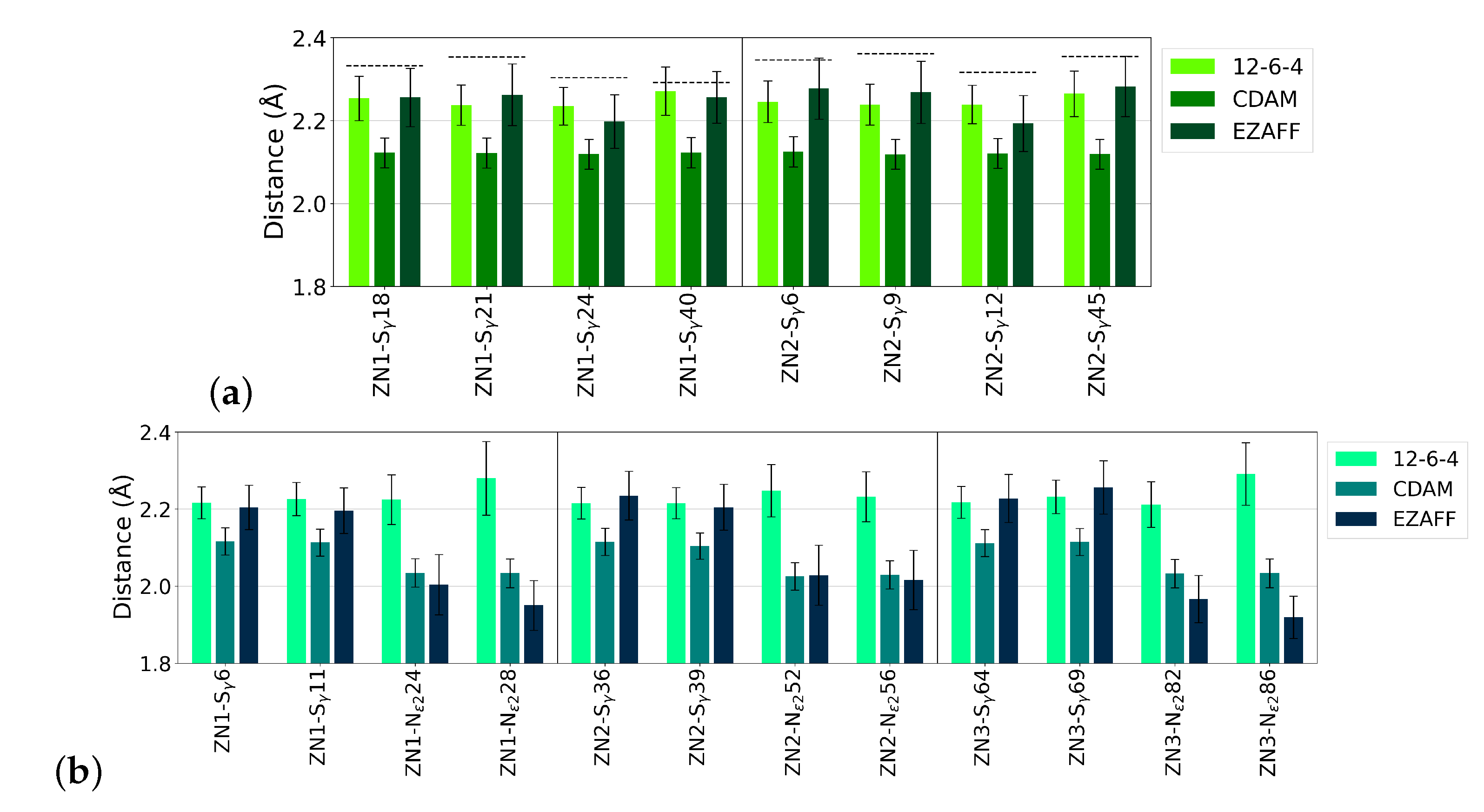
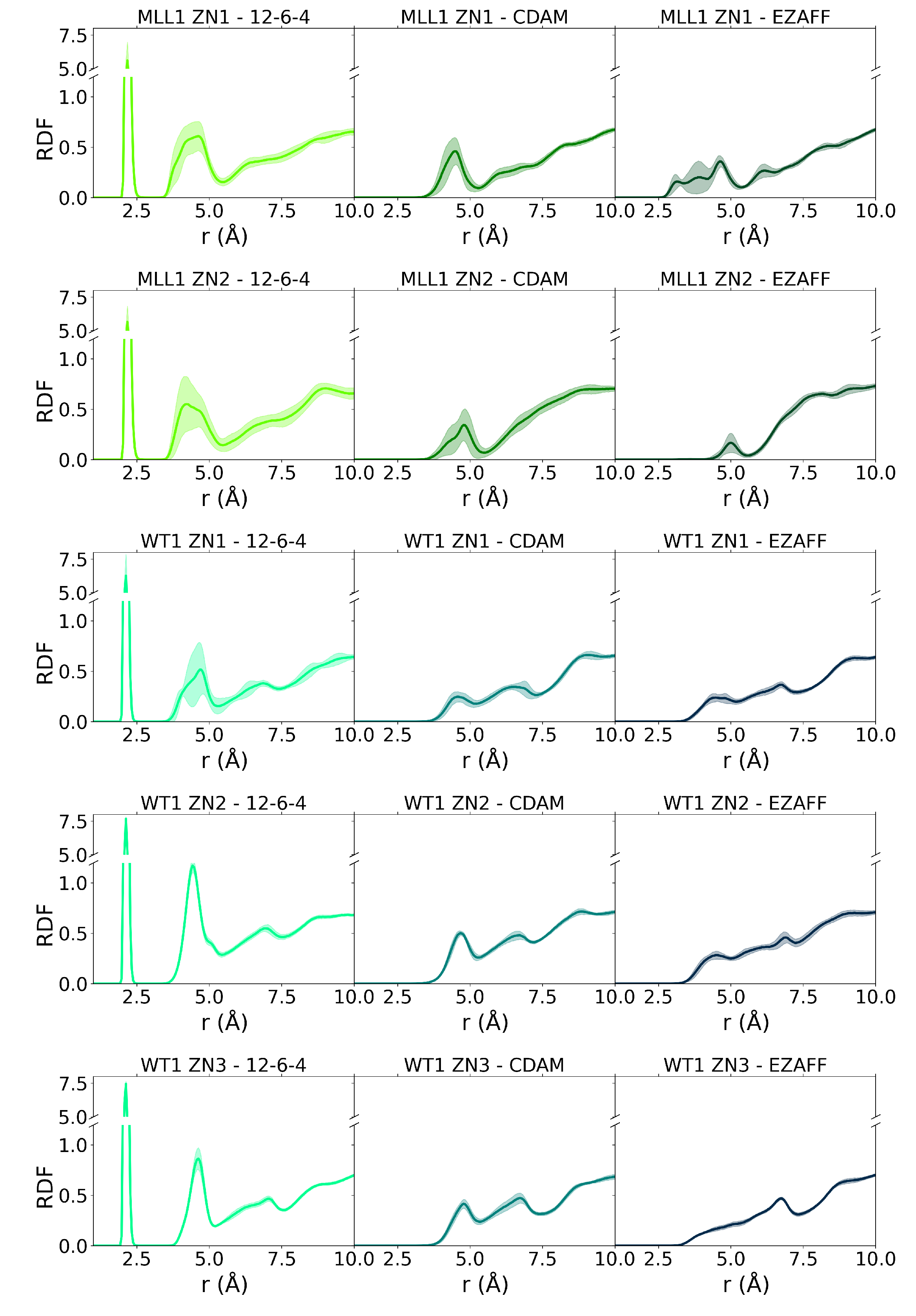
3.4. Zn Coordination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDAM | cationic dummy atom model |
| EZAFF | extended zinc AMBER force field |
| MD | molecular dynamics |
| CN | coordination number |
References
- Lu, Y.; Yeung, N.; Sieracki, N.; Marshall, N.M. Design of Functional Metalloproteins. Nature 2009, 460, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahrenkamp, H. Why does nature use zinc-a personal view. Dalton Trans. 2007, 42, 4751–4759. [Google Scholar] [CrossRef]
- Gropper, S.S.; Smith, J.L.; Carr, T.P. Advanced Nutrition and Human Metabolism; Wadsworth: Belmont, CA, USA, 2021. [Google Scholar]
- Cherasse, Y.; Urade, Y. Dietary Zinc Acts as a Sleep Modulator. Int. J. Mol. Sci. 2017, 18, 2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markov, D.; Naryshkina, T.; Mustaev, A.; Severinov, K. A zinc-binding site in the largest subunit of DNA-dependent RNA polymerase is involved in enzyme assembly. Genes Dev. 1999, 13, 2439–2448. [Google Scholar] [CrossRef] [Green Version]
- Rich, A.M.; Bombarda, E.; Schenk, A.D.; Lee, P.E.; Cox, E.H.; Spuches, A.M.; Hudson, L.D.; Kieffer, B.; Wilcox, D.E. Thermodynamics of Zn2+ Binding to Cys2His2 and Cys2HisCys Zinc Fingers and a Cys4 Transcription Factor Site. J. Am. Chem. Soc. 2012, 134, 10405–10418. [Google Scholar] [CrossRef]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar] [CrossRef]
- Schöne, S.; Jurk, M.; Helabad, M.B.; Dror, I.; Lebars, I.; Kieffer, B.; Imhof, P.; Rohs, R.; Vingron, M.; Thomas-Chollier, M.; et al. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat. Commun. 2016, 7, 12621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Merz, K.M. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, W.N.; Sträter, N. Recent Advances in Zinc Enzymology. Chem. Rev. 1996, 96, 2375–2434. [Google Scholar] [CrossRef]
- Laitaoja, M.; Valjakka, J.; Jänis, J. Zinc Coordination Spheres in Protein Structures. Inorg. Chem. 2013, 52, 10983–10991. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Piquemal, J.P.; Chaudret, R.; Reinhardt, P.; Ren, P. Polarizable Molecular Dynamics Simulation of Zn(II) in Water Using the AMOEBA Force Field. J. Chem. Theory Comput. 2010, 6, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgrading and Validation of the AMBER Force Field for Histidine and Cysteine Zinc(II)-Binding Residues in Sites with Four Protein Ligands. J. Chem. Inf. Model. 2019, 59, 3803–3816. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgraded AMBER Force Field for Zinc-Binding Residues and Ligands for Predicting Structural Properties and Binding Affinities in Zinc-Proteins. ACS Omega 2020, 5, 15301–15310. [Google Scholar] [CrossRef]
- Melse, O.; Antes, I.; Kaila, V.R.I.; Zacharias, M. Benchmarking biomolecular force field-based Zn2+ for mono- and bimetallic ligand binding sites. J. Comput. Chem. 2022, 44, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. Taking into Account the Ion-induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Song, L.F.; Li, P.; Merz, K.M. Systematic Parametrization of Divalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Theory Comput. 2020, 16, 4429–4442. [Google Scholar] [CrossRef] [PubMed]
- Song, L.F.; Sengupta, A.; Merz, K.M. Thermodynamics of Transition Metal Ion Binding to Proteins. J. Am. Chem. Soc. 2020, 142, 6365–6374. [Google Scholar] [CrossRef]
- Panteva, M.T.; Giambaşu, G.M.; York, D.M. Force Field for Mg2+, Mn2+, Zn2+, and Cd2+ Ions That Have Balanced Interactions with Nucleic Acids. J. Phys. Chem. B 2015, 119, 15460–15470. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Molecular Dynamics Simulations: Difficulties, Solutions and Strategies for Treating Metalloenzymes. In Challenges and Advances in Computational Chemistry and Physics; Springer Netherlands: Dordrecht, The Netherlands, 2010; pp. 299–330. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Li, P.; Merz, K.M. Extended Zinc AMBER Force Field (EZAFF). J. Chem. Theory Comput. 2018, 14, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M.J. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ryde, U. Comparison of Methods to Obtain Force-Field Parameters for Metal Sites. J. Chem. Theory Comput. 2011, 7, 2452–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.P. Novel Zinc Protein Molecular Dynamics Simulations: Steps toward Antiangiogenesis for Cancer Treatment. J. Mol. Model. 1999, 5, 196–202. [Google Scholar] [CrossRef]
- Pang, Y.P.; Xu, K.; Yazal, J.E.; Prendergas, F.G. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. Publ. Protein Soc. 2000, 9, 1857–1865. [Google Scholar]
- Pang, Y.P. Successful molecular dynamics simulation of two zinc complexes bridged by a hydroxide in phosphotriesterase using the cationic dummy atom method. Proteins 2001, 45, 183–189. [Google Scholar] [CrossRef]
- Duarte, F.; Bauer, P.; Barrozo, A.; Amrein, B.A.; Purg, M.; Aqvist, J.; Kamerlin, S.C.L. Force Field Independent Metal Parameters Using a Nonbonded Dummy Model. J. Phys. Chem. B 2014, 118, 4351–4362. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Tan, T. Rational Design of Methodology-Independent Metal Parameters Using a Nonbonded Dummy Model. J. Chem. Theory Comput. 2016, 12, 3250–3260. [Google Scholar] [CrossRef] [Green Version]
- Tomić, A.; Horvat, G.; Ramek, M.; Agić, D.; Brkić, H.; Tomić, S. New Zinc Ion Parameters Suitable for Classical MD Simulations of Zinc Metallopeptidases. J. Chem. Inf. Model. 2019, 59, 3437–3453. [Google Scholar] [CrossRef]
- Jardin, C.; Chaves, G.; Musset, B. Assessing Structural Determinants of Zn2+ Binding to Human HV1 via Multiple MD Simulations. Biophys. J. 2020, 118, 1221–1233. [Google Scholar] [CrossRef]
- MacDermott-Opeskin, H.; McDevitt, C.A.; O’Mara, M.L. Comparing Nonbonded Metal Ion Models in the Divalent Cation Binding Protein PsaA. J. Chem. Theory Comput. 2020, 16, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Zhang, X.; Zheng, Y.; Wilson, G.G.; Cheng, X. Denys-Drash syndrome associated WT1 glutamine 369 mutants have altered sequence-preferences and altered responses to epigenetic modifications. Nucleic Acids Res. 2016, 44, 10165–10176. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, K.; Lei, M.; Yang, A.; Li, Y.; Hughes, T.R.; Min, J. DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Structure 2018, 26, 85–95.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, H.; Cheng, X. Wilms Tumor Protein (WT1) ZnF2-4 in Complex with DNA. 2016. Available online: https://ftp.wwpdb.org/pub/pdb/validation_reports/kl/5kl2/5kl2_full_validation.pdf.gz (accessed on 28 January 2023). [CrossRef]
- Bian, C.; Tempel, W.; Chao, X.; Walker, J.; Bountra, C.; Weigelt, J.; Arrowsmith, C.; Edwards, A.; Min, J. Crystal Structure of MLL CXXC Domain in Complex with a CpG DNA. 2014. Available online: https://ftp.wwpdb.org/pub/pdb/validation_reports/nw/4nw3/4nw3_full_validation.pdf.gz (accessed on 28 January 2023). [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Martínez-Rosell, G.; Giorgino, T.; de Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Go ni, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. AMBER 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Pang, Y.P. Zinc Protein Simulations Using the Cationic Dummy Atom (CaDA) Method. Available online: https://www.mayo.edu/research/labs/computer-aided-molecular-design/projects/zinc-protein-simulations-using-cationic-dummy-atom-cada-approach (accessed on 28 January 2023).
- Li, P.; Merz, K.M. Building Bonded Model for A Ligand Binding Metalloprotein with MCPB.py. Available online: http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm (accessed on 28 January 2023).
- Frisch, M.E.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, M. Atomic Resolution Insight into Sac7d Protein Binding to DNA and Associated Global Changes by Molecular Dynamics Simulations. Angew. Chem. (Int. Ed. Engl.) 2019, 58, 5967–5972. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreini, C.; Cavallaro, G.; Lorenzini, S. FindGeo: A tool for determining metal coordination geometry. Bioinformatics 2012, 28, 1658–1660. [Google Scholar] [CrossRef] [Green Version]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Joosten, R.P.; Salzemann, J.; Bloch, V.; Stockinger, H.; Berglund, A.C.; Blanchet, C.; Bongcam-Rudloff, E.; Combet, C.; Da Costa, A.L.; Deleage, G.; et al. PDB_REDO: Automated re-refinement of X-ray structure models in the PDB. J. Appl. Crystallogr. 2009, 42, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, N.W.; Janowski, P.A.; Swails, J.M.; Nguyen, H.; Richardson, J.S.; Case, D.A.; Adams, P.D. Improved chemistry restraints for crystallographic refinement by integrating the Amber force field into Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 51–62. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordination Geometry (CN) | 12-6-4 | CDAM | EZAFF |
|---|---|---|---|
| MLL1 ZN1, Regular Tetrahedron (4) | |||
| Tetrahedron (4) | 0.82 | 100.00 | 99.16 |
| Trigonal bipyramid with a vacancy (axial) (4) | – | – | 0.25 |
| Trigonal bipyramid (5) | 13.26 | – | – |
| Square pyramid (5) | 0.11 | – | – |
| Trigonal prism with a vacancy (5) | 0.02 | – | – |
| Octahedron (6) | 78.18 | – | – |
| Pentagonal bipyramid with a vacancy (equatorial) (6) | 7.30 | – | – |
| Irregular geometry | 0.32 | – | 0.60 |
| MLL1 ZN2, Distorted Tetrahedron (4) | |||
| Tetrahedron (4) | 0.26 | 100.00 | 55.18 |
| Trigonal bipyramid with a vacancy (axial) (4) | – | – | 28.14 |
| Square pyramid with a vacancy (equatorial) (4) | – | – | 15.30 |
| Trigonal bipyramid with a vacancy (equatorial) (4) | – | – | 1.39 |
| Trigonal bipyramid (5) | 13.54 | – | – |
| Square pyramid (5) | 0.02 | – | – |
| Octahedron (6) | 82.12 | – | – |
| Pentagonal bipyramid with a vacancy (equatorial) (6) | 4.02 | – | – |
| Irregular geometry | 0.04 | – | – |
| WT1 ZN1, Regular Tetrahedron (4) | |||
| Tetrahedron (4) | – | 100.00 | 99.12 |
| Trigonal bipyramid with a vacancy (axial) (4) | – | – | 0.88 |
| Square pyramid (5) | 0.02 | – | – |
| Octahedron (6) | 96.79 | – | – |
| Pentagonal bipyramid with a vacancy (equatorial) (6) | 3.17 | – | – |
| Octahedron, face monocapped with a vacancy (capped face) (6) | 0.02 | – | – |
| WT1 ZN2, Regular Tetrahedron (4) | |||
| Tetrahedron (4) | – | 99.98 | 99.96 |
| Trigonal bipyramid with a vacancy (axial) (4) | – | 0.02 | 0.04 |
| Octahedron (6) | 99.12 | – | – |
| Pentagonal bipyramid with a vacancy (equatorial) (6) | 0.86 | – | – |
| Octahedron, face-monocapped with a vacancy (capped face) (6) | 0.02 | – | – |
| WT1 ZN3, Regular Tetrahedron (4) | |||
| Tetrahedron (4) | – | 99.98 | 96.81 |
| Trigonal bipyramid with a vacancy (axial) (4) | – | 0.02 | 3.19 |
| Octahedron (6) | 92.58 | – | – |
| Pentagonal bipyramid with a vacancy (equatorial) (6) | 7.42 | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkenandt, S.; Imhof, P. Comparison of Empirical Zn2+ Models in Protein–DNA Complexes. Biophysica 2023, 3, 214-230. https://doi.org/10.3390/biophysica3010014
Volkenandt S, Imhof P. Comparison of Empirical Zn2+ Models in Protein–DNA Complexes. Biophysica. 2023; 3(1):214-230. https://doi.org/10.3390/biophysica3010014
Chicago/Turabian StyleVolkenandt, Senta, and Petra Imhof. 2023. "Comparison of Empirical Zn2+ Models in Protein–DNA Complexes" Biophysica 3, no. 1: 214-230. https://doi.org/10.3390/biophysica3010014
APA StyleVolkenandt, S., & Imhof, P. (2023). Comparison of Empirical Zn2+ Models in Protein–DNA Complexes. Biophysica, 3(1), 214-230. https://doi.org/10.3390/biophysica3010014