

Nuclear Motion Is Classical: Spectra of Hydrogen Chloride and Ammonia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
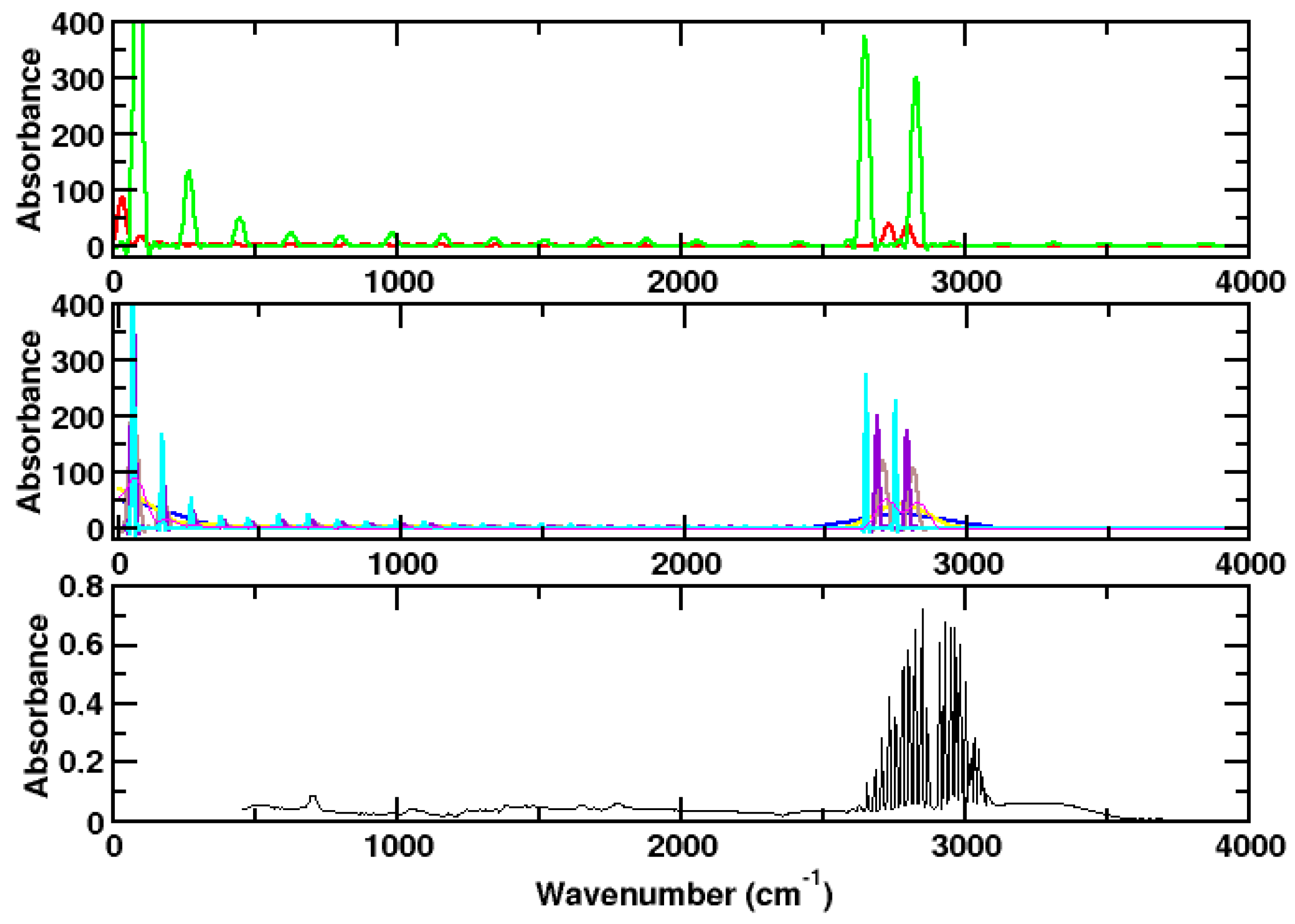
:1. Introduction
2. Methods
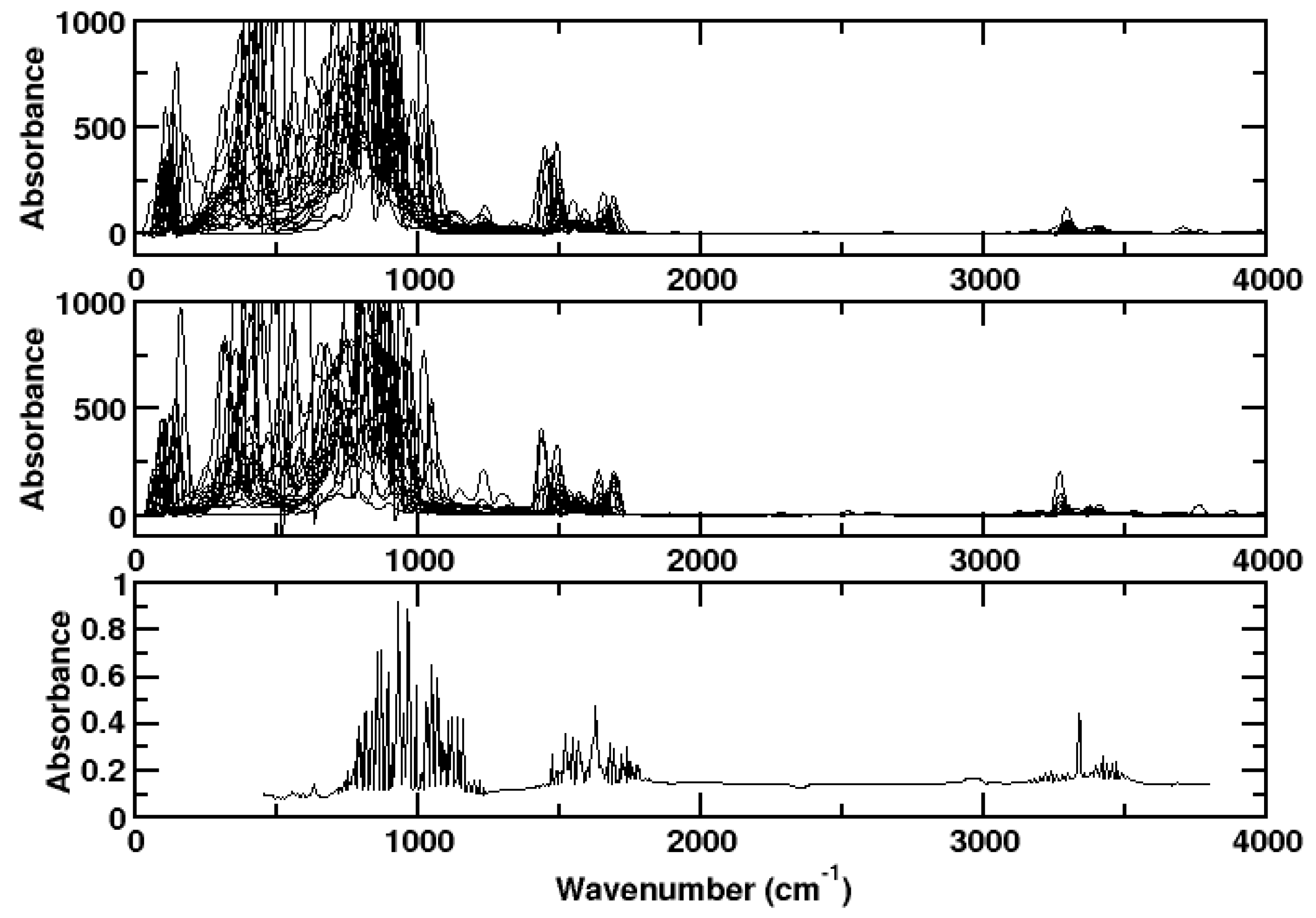
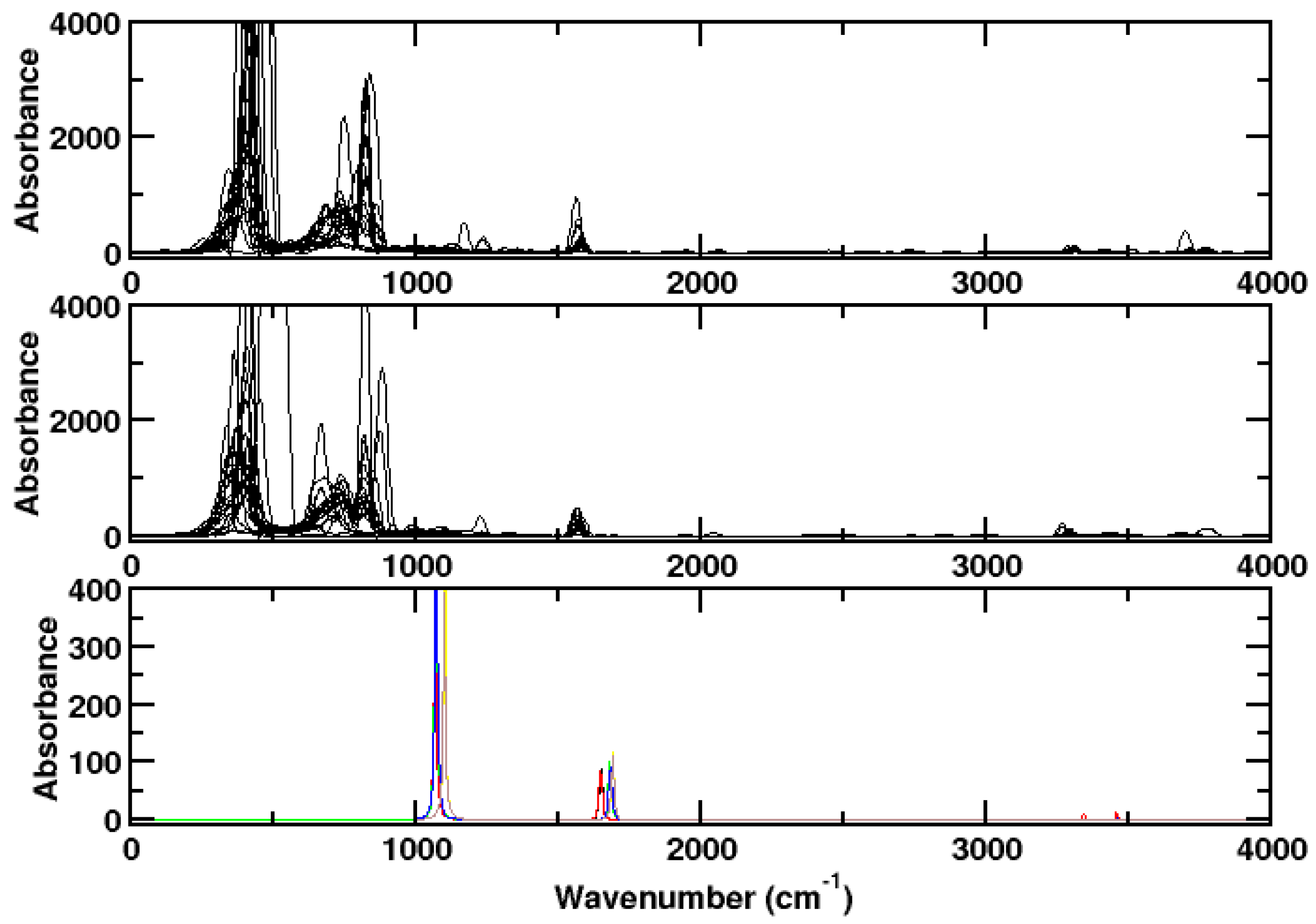
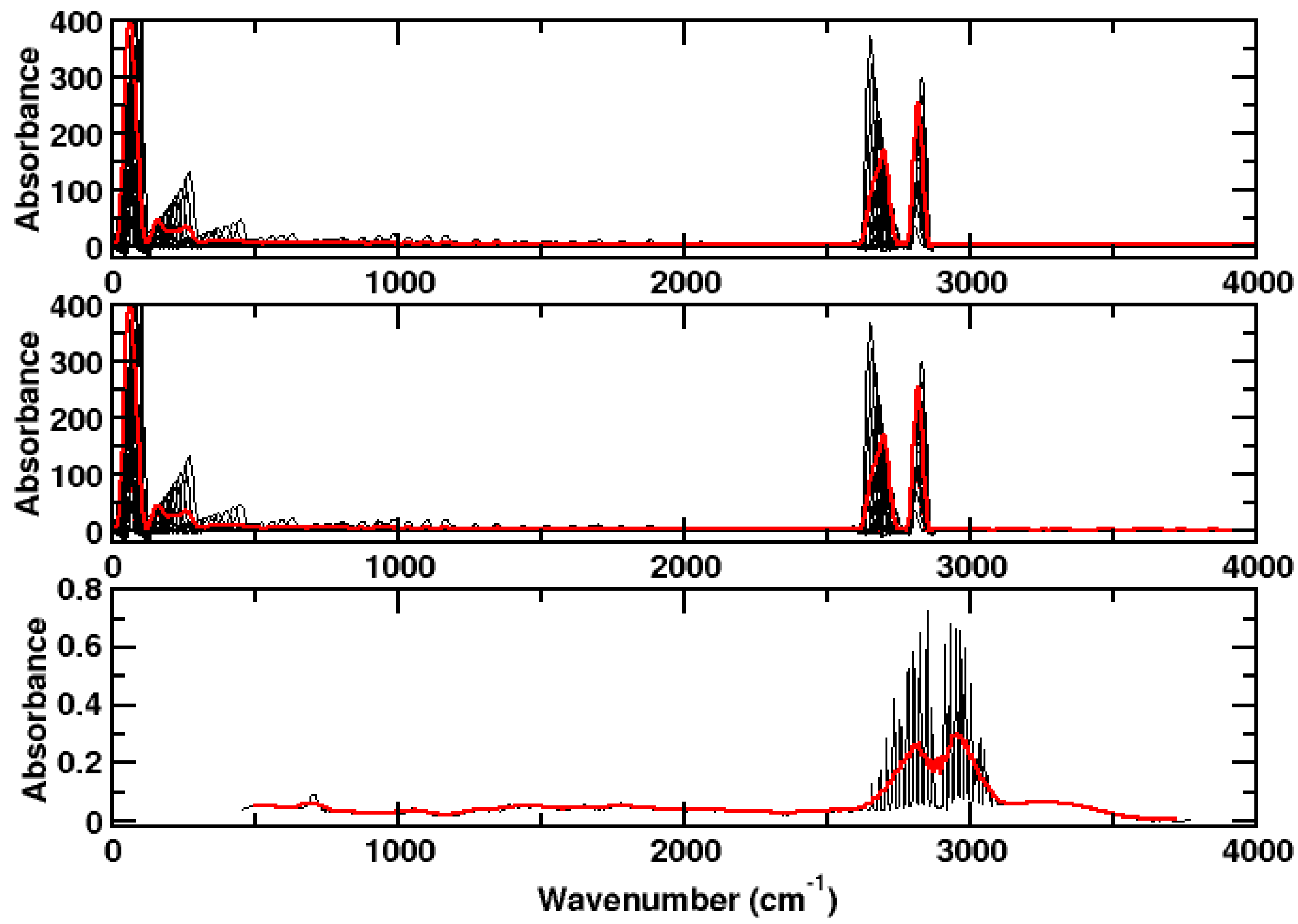
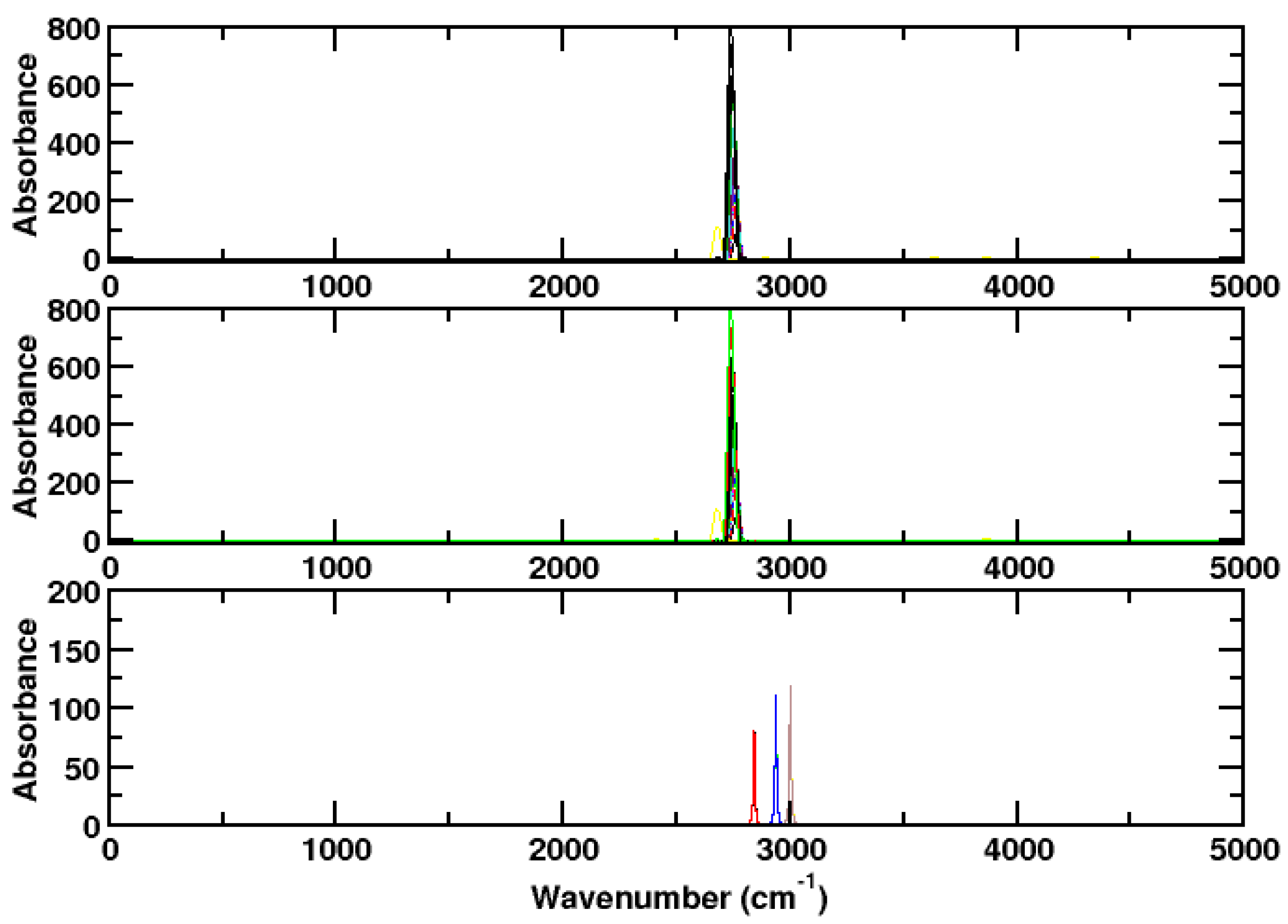
3. Results and Discussion
3.1. Ammonia
3.2. Hydrogen Chloride
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIMD | Ab Initio Molecular Dynamics |
| BLYP | Becke–Lee–Yang–Parr density functional |
| B3LYP | Hybrid functional based on BLYP |
| B2PLYP | Double-hybrid functional based on B3LYP and MP2 |
| CPMD | Car–Parrinello Molecular Dynamics |
| -D3: | With dispersion correction |
| DFT | Density functional theory |
| MP2 | Moeller–Plesset second-order perturbation theory |
References
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Frank, I.; Genuit, S.; Matz, F.; Oschinski, H. Ammonia, water, and hydrogen: Can nuclear motion be described classically? Int. J. Quantum Chem. 2020, 120, e26142. [Google Scholar] [CrossRef]
- Frank, I. Classical motion of the nuclei in a molecule: A concept without alternatives. Chem. Select 2020, 5, 1872. [Google Scholar] [CrossRef]
- Büchel, R.C.; Rudolph, D.A.; Frank, I. Deterministic quantum mechanics: The role of the Maxwell-Boltzmann distribution. Int. J. Quantum Chem. 2021, 121, e26555. [Google Scholar] [CrossRef]
- Frank, I. Classical nuclear motion: Comparison to approaches with quantum mechanical nuclear motion. Hydrogen 2023, 4, 11–21. [Google Scholar] [CrossRef]
- Yurchenko, S.N.; Barber, R.J.; Tennyson, J.; Thiel, W.; Jensen, P. Towards efficient refinement of molecular potential energy surfaces: Ammonia as a case study. J. Mol. Spectrosc. 2011, 268, 123. [Google Scholar] [CrossRef]
- Marquardt, R.; Sagui, K.; Zheng, J.; Thiel, W.; Luckhaus, D.; Yurchenko, S.N.; Mariotti, F.; Quack, M. Global analytical potential energy surface for the electronic ground state of NH3 from high level ab initio calculations. J. Phys. Chem. A 2013, 117, 7502. [Google Scholar] [CrossRef]
- Fabri, C.; Marquardt, R.; Csaszar, A.G.; Quack, M. Controlling tunneling in ammonia isotopomers. J. Chem. Phys. 2019, 150, 014102. [Google Scholar] [CrossRef]
- Hutter, J.; Alavi, A.; Deutsch, T.; Bernasconi, M.; Goedecker, S.; Marx, D.; Tuckerman, M.; Parrinello, M. CPMD Version 4.3. Copyright IBM Corp 1990–2015. Copyright MPI für Festkörperforschung Stuttgart 1997–2001. Available online: https://github.com/CPMD-code/CPMD/releases/tag/4.3 (accessed on 11 May 2023).
- Grimme, S. Accurate Description of van der Waals Complexes by Density Functional Theory Including Empirical Corrections. J. Comput. Chem. 2004, 25, 1463. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Boero, M.; Parrinello, M.; Terakura, K.; Weiss, H. Car-Parrinello study of Ziegler-Natta heterogeneous catalysis: Stability and destabilization problems of the active site models. Mol. Phys. 2002, 100, 2935–2940. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS—A free analyzer and visualizer for monte carlo and molecular dynamics trajectories. J. Chem. Inf. Model. 2011, 51, 2007. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS—A free analyzer for trajectories from molecular simulation. J. Chem. Phys. 2020, 152, 164105. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Brehm, M.; Fligg, R.; Vöhringer, P.; Kirchner, B. Computing vibrational spectra from ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 6608. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Brehm, M.; Kirchner, B. Voronoi dipole moments for the simulation of bulk phase vibrational spectra. Phys. Chem. Chem. Phys. 2015, 17, 3207. [Google Scholar] [CrossRef] [PubMed]
- Marzari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847. [Google Scholar] [CrossRef]
- Ikeda, T.; Boero, M. Role of van der Waals corrections in first principles simulations of alkali metal ions in aqueous solutions. J. Chem. Phys. 2015, 143, 194510. [Google Scholar] [CrossRef] [PubMed]
- Silvestrelli, P.L.; Ambrosetti, A. Van der Waals interactions in DFT using Wannier functions without empirical parameters. J. Chem. Phys. 2019, 150, 164109. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Becke, A. A New Mixing of Hartree-Fock and Local-Density Functional Theories. J. Chem. Phys. 1993, 98, 1372. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- NIST Chemistry WebBook. Available online: https://webbook.nist.gov/ (accessed on 30 December 2022).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, I. Nuclear Motion Is Classical: Spectra of Hydrogen Chloride and Ammonia. Hydrogen 2023, 4, 287-294. https://doi.org/10.3390/hydrogen4020020
Frank I. Nuclear Motion Is Classical: Spectra of Hydrogen Chloride and Ammonia. Hydrogen. 2023; 4(2):287-294. https://doi.org/10.3390/hydrogen4020020
Chicago/Turabian StyleFrank, Irmgard. 2023. "Nuclear Motion Is Classical: Spectra of Hydrogen Chloride and Ammonia" Hydrogen 4, no. 2: 287-294. https://doi.org/10.3390/hydrogen4020020
APA StyleFrank, I. (2023). Nuclear Motion Is Classical: Spectra of Hydrogen Chloride and Ammonia. Hydrogen, 4(2), 287-294. https://doi.org/10.3390/hydrogen4020020