
Advancements in Understanding and Treating NAFLD: A Comprehensive Review of Metabolic-Associated Fatty Liver Disease and Emerging Therapies

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Epidemiology
Prevalence
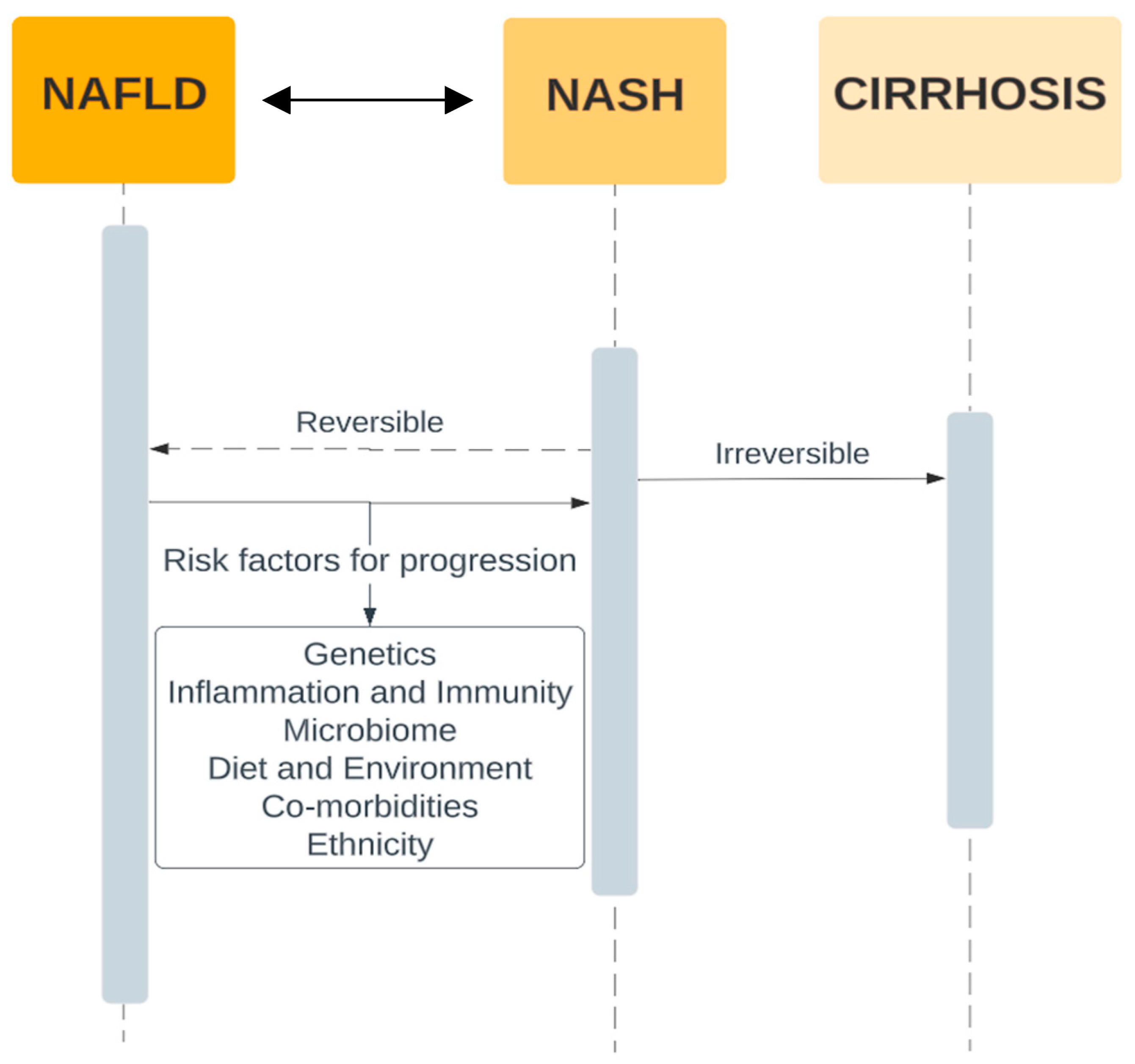
3. Pathogenesis of NAFLD and Progression to Fibrosis
3.1. Adiposity—Liver Axis and Insulin Resistance
3.2. Inflammatory Pathways
3.3. Gut/Liver Axis and Gut Microbiome
3.4. Dietary and Environmental Factors
3.5. Genetics
4. Diagnosis
5. Treatments
6. Emerging Therapeutic Options
6.1. Semaglutide
6.2. Obeticholic Acid
6.3. Lanifibranor
6.4. Arachidyl Amido Cholanoic Acid
6.5. Resmetirom
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janssen, A.; Grobbee, D.E.; Dendale, P. Non-alcoholic fatty liver disease, a new and growing risk indicator for cardiovascular disease. Eur. J. Prev. Cardiol. 2020, 27, 1059–1063. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Parikh, R.M.; Mohan, V. Changing definitions of metabolic syndrome. Indian J. Endocrinol. Metab. 2012, 16, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.Y.; Yuen, M.F.; Seto, W.K. Letter regarding “A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement”. J. Hepatol. 2020, 73, 1573–1574. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.E.; Koh, T.J.L.; Tang, A.S.P.; Quek, J.; Yong, J.N.; Tay, P.; Tan, D.J.H.; Lim, W.H.; Lin, S.Y.; Huang, D.; et al. Global Prevalence and Clinical Characteristics of Metabolic-associated Fatty Liver Disease: A Meta-Analysis and Systematic Review of 10 739 607 Individuals. J. Clin. Endocrinol. Metab. 2022, 107, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Xian, Y.X.; Weng, J.P.; Xu, F. MAFLD vs. NAFLD: Shared features and potential changes in epidemiology, pathophysiology, diagnosis, and pharmacotherapy. Chin. Med. J. 2020, 134, 8–19. [Google Scholar] [CrossRef]
- Shaker, M. Liver transplantation for nonalcoholic fatty liver disease: New challenges and new opportunities. World J. Gastroenterol. 2014, 20, 5320–5330. [Google Scholar] [CrossRef]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Wong, G.L.-H.; Woo, J.; Abrigo, J.M.; Chan, C.K.-M.; Shu, S.S.-T.; Leung, J.K.-Y.; Chim, A.M.-L.; Kong, A.P.-S.; Lui, G.C.-Y.; et al. Impact of the New Definition of Metabolic Associated Fatty Liver Disease on the Epidemiology of the Disease. Clin. Gastroenterol. Hepatol. 2021, 19, 2161–2171.e5. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R. Trends in the Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in the United States, 2011–2018. Clin. Gastroenterol. Hepatol. 2022, 20, e610–e613. [Google Scholar] [CrossRef]
- Ciardullo, S.; Perseghin, G. Prevalence of NAFLD, MAFLD and associated advanced fibrosis in the contemporary United States population. Liver Int. 2021, 41, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, X.; Li, G.; Wong, G.L.-H.; Wong, V.W.-S. Epidemiology and Clinical Outcomes of Metabolic (Dysfunction)-associated Fatty Liver Disease. J. Clin. Transl. Hepatol. 2021, 9, 972–982. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.; Salam, R.A.; Lassi, Z.S.; Das, J.K. The Intertwined Relationship between Malnutrition and Poverty. Front. Public Health 2020, 8, 453. [Google Scholar] [CrossRef]
- Jenssen, B.P.; Kelly, M.K.; Powell, M.; Bouchelle, Z.; Mayne, S.L.; Fiks, A.G. COVID-19 and Changes in Child Obesity. Pediatrics 2021, 147, e2021050123. [Google Scholar] [CrossRef]
- Noonan, R.J. Poverty, Weight Status, and Dietary Intake among UK Adolescents. Int. J. Environ. Res. Public Health 2018, 15, 1224. [Google Scholar] [CrossRef]
- Hoffmann, C.; Gerber, P.A.; Cavelti-Weder, C.; Licht, L.; Kotb, R.; Al Dweik, R.; Cherfane, M.; Bornstein, S.R.; Perakakis, N. Liver, NAFLD and COVID-19. Horm. Metab. Res. 2022, 54, 522–531. [Google Scholar] [CrossRef]
- Fujii, H.; Nakamura, N.; Fukumoto, S.; Kimura, T.; Nakano, A.; Nadatani, Y.; Tauchi, Y.; Nishii, Y.; Takashima, S.; Kamada, Y.; et al. Lifestyle changes during the coronavirus disease 2019 pandemic impact metabolic dysfunction-associated fatty liver disease. Liver Int. 2022, 42, 995–1004. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef]
- Da Silva, H.E.; Arendt, B.M.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Allard, J.P. A cross-sectional study assessing dietary intake and physical activity in Canadian patients with nonalcoholic fatty liver disease vs healthy controls. J. Acad. Nutr. Diet. 2014, 114, 1181–1194. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Liu, Q.; Bengmark, S.; Qu, S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2010, 9, 42. [Google Scholar] [CrossRef]
- Shulman, G.I. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 2014, 371, 2237–2238. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Stojsavljević, S.; Gomerčić Palčić, M.; Virović Jukić, L.; Smirčić Duvnjak, L.; Duvnjak, M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 18070–18091. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Das, S.K.; Balakrishnan, V. Role of cytokines in the pathogenesis of non-alcoholic Fatty liver disease. Indian J. Clin. Biochem. 2011, 26, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Langendijk, P.S.; Schut, F.; Jansen, G.J.; Raangs, G.C.; Kamphuis, G.R.; Wilkinson, M.H.; Welling, G.W. Quantitative fluorescence in situ hybridization of Bifidobacterium spp. with genus-specific 16S rRNA-targeted probes and its application in fecal samples. Appl. Environ. Microbiol. 1995, 61, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Dumas, M.-E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef]
- Herrmann, E.; de Lédinghen, V.; Cassinotto, C.; Chu, W.C.W.; Leung, V.Y.F.; Ferraioli, G.; Filice, C.; Castera, L.; Vilgrain, V.; Ronot, M.; et al. Assessment of biopsy-proven liver fibrosis by two-dimensional shear wave elastography: An individual patient data-based meta-analysis. Hepatology 2018, 67, 260–272. [Google Scholar] [CrossRef]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef] [PubMed]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef] [PubMed]
- De Wit, N.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; de Vogel-van den Bosch, J.; Kleerebezem, M.; Müller, M.; van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G589–G599. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Sarfati, G.; Nubret, E.; Kapel, N.; Waligora-Dupriet, A.-J.; Bergheim, I.; Cynober, L.; De-Bandt, J.-P. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin. Nutr. 2016, 35, 175–182. [Google Scholar] [CrossRef]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int. J. Hepatol. 2014, 2014, 560620. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Cohney, S.; Pinach, S.; Saba, F.; Gambino, R. Emerging Liver-Kidney Interactions in Nonalcoholic Fatty Liver Disease. Trends Mol. Med. 2015, 21, 645–662. [Google Scholar] [CrossRef]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andrés-Hernando, A.; Ishimoto, T.; Sánchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.; Tümer, N.; Gao, Y.; Cheng, K.-Y.; Scarpace, P.J. Prevention and reversal of diet-induced leptin resistance with a sugar-free diet despite high fat content. Br. J. Nutr. 2011, 106, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.-Y.; Johnson, R.J.; Scarpace, P.J.; Patel, C.; Sugimoto, K.; Douard, V.; Shah, A.; et al. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1370–R1375. [Google Scholar] [CrossRef] [PubMed]
- Tarnoki, A.D.; Tarnoki, D.L.; Bata, P.; Littvay, L.; Osztovits, J.; Jermendy, G.; Karlinger, K.; Lannert, A.; Preda, I.; Kiss, R.G.; et al. Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: A twin study. Liver Int. 2012, 32, 1287–1293. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef]
- Loomba, R.; Schork, N.; Chen, C.-H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- Wagenknecht, L.E.; Scherzinger, A.L.; Stamm, E.R.; Hanley, A.J.; Norris, J.M.; Chen, Y.-D.I.; Bryer-Ash, M.; Haffner, S.M.; Rotter, J.I. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity 2009, 17, 1240–1246. [Google Scholar] [CrossRef]
- Palmer, N.D.; Musani, S.K.; Yerges-Armstrong, L.M.; Feitosa, M.F.; Bielak, L.F.; Hernaez, R.; Kahali, B.; Carr, J.J.; Harris, T.B.; Jhun, M.A.; et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 2013, 58, 966–975. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744 e7. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yao, S. Expression, purification of herpes simplex virus type 1 US11 protein, and production of US11 polyclonal antibody. Virol. J. 2011, 8, 490. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Mallardi, P.; Gianotti, T.F.; Burgueño, A.L.; Martino, J.S.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230 e6. [Google Scholar] [CrossRef]
- Kim, U.; Kim, N.; Shin, H.Y. Modeling Non-Alcoholic Fatty Liver Disease (NAFLD) Using “Good-Fit” Genome-Editing Tools. Cells 2020, 9, 2572. [Google Scholar] [CrossRef]
- Petta, S.; Miele, L.; Bugianesi, E.; Cammà, C.; Rosso, C.; Boccia, S.; Cabibi, D.; Di Marco, V.; Grimaudo, S.; Grieco, A.; et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e87523. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Tsang, S.W.; Hui, A.Y.; Chan, A.; Choi, P.C.; Chim, A.M.; Chu, S.; Chan, F.K.; Sung, J.J.; et al. Metabolic and histological features of non-alcoholic fatty liver disease patients with different serum alanine aminotransferase levels. Aliment. Pharmacol. Ther. 2009, 29, 387–396. [Google Scholar] [CrossRef]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281 e4. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridi, M.; Cholongitas, E. Diagnosis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Concepts. Curr. Pharm. Des. 2018, 24, 4574–4586. [Google Scholar] [CrossRef] [PubMed]
- Khov, N.; Sharma, A.; Riley, T.R. Bedside ultrasound in the diagnosis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 6821–6825. [Google Scholar] [CrossRef] [PubMed]
- Kinner, S.; Reeder, S.B.; Yokoo, T. Quantitative Imaging Biomarkers of NAFLD. Dig. Dis. Sci. 2016, 61, 1337–1347. [Google Scholar] [CrossRef]
- Loomba, R. Role of imaging-based biomarkers in NAFLD: Recent advances in clinical application and future research directions. J. Hepatol. 2018, 68, 296–304. [Google Scholar] [CrossRef]
- Li, Q.; Dhyani, M.; Grajo, J.R.; Sirlin, C.; Samir, A.E. Current status of imaging in nonalcoholic fatty liver disease. World J. Hepatol. 2018, 10, 530–542. [Google Scholar] [CrossRef]
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wong, V.W.-S.; Peleg, N.; et al. Association between Fibrosis Stage and Outcomes of Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625 e12. [Google Scholar] [CrossRef]
- Tsai, E.; Lee, T.P. Diagnosis and Evaluation of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis, Including Noninvasive Biomarkers and Transient Elastography. Clin. Liver Dis. 2018, 22, 73–92. [Google Scholar] [CrossRef]
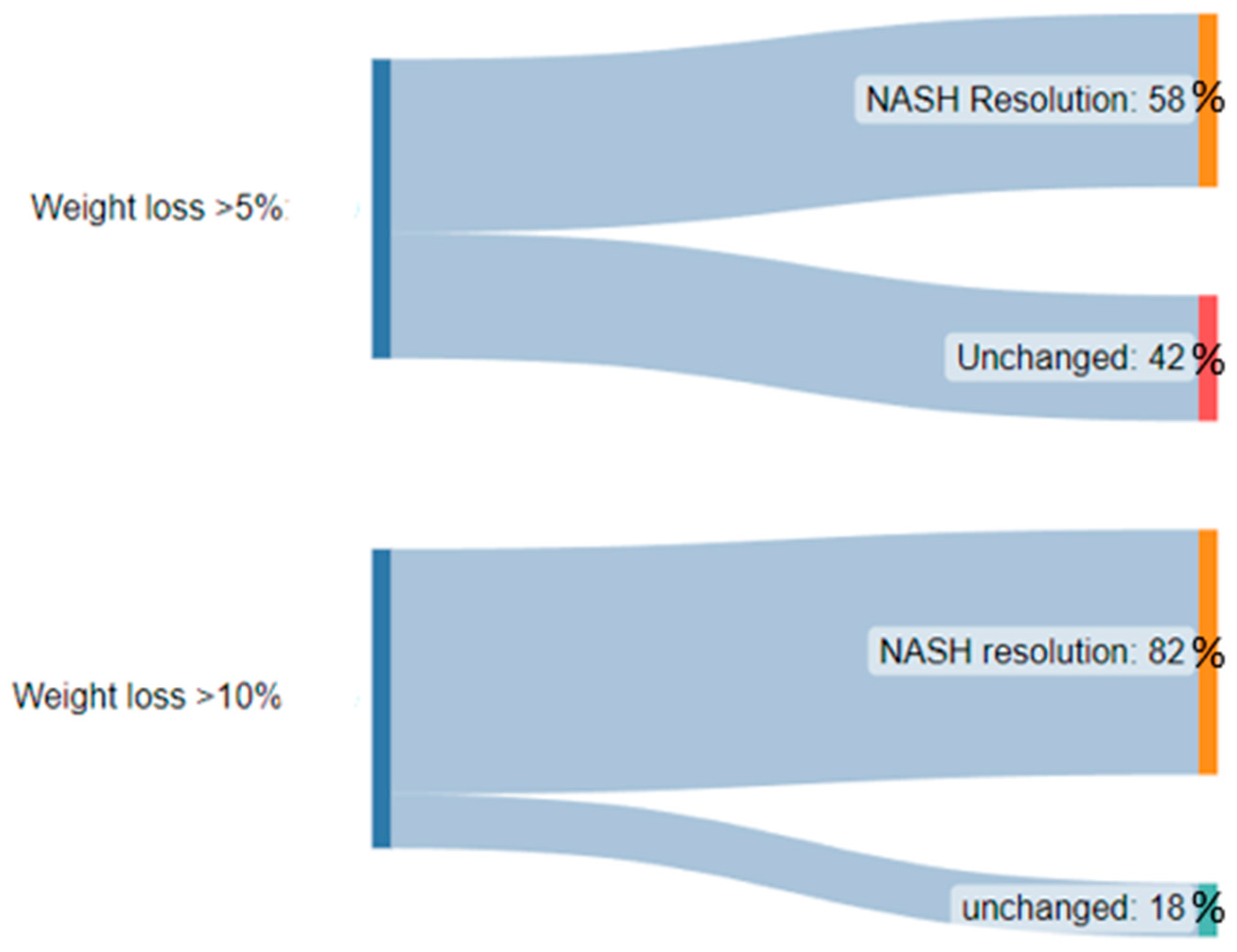
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gómez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef]
- Hannah, W.N.; Harrison, S.A., Jr. Effect of Weight Loss, Diet, Exercise, and Bariatric Surgery on Nonalcoholic Fatty Liver Disease. Clin. Liver Dis. 2016, 20, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014, 129 (Suppl. S2), S102–S138. [Google Scholar] [CrossRef]
- Bower, G.; Toma, T.; Harling, L.; Jiao, L.R.; Efthimiou, E.; Darzi, A.; Athanasiou, T.; Ashrafian, H. Bariatric Surgery and Non-Alcoholic Fatty Liver Disease: A Systematic Review of Liver Biochemistry and Histology. Obes. Surg. 2015, 25, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Tapia, N.C.; Tellez-Avila, F.I.; Barrientos-Gutierrez, T.; Mendez-Sanchez, N.; Lizardi-Cervera, J.; Uribe, M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst. Rev. 2010, 2010, CD007340. [Google Scholar] [CrossRef]
- Mathurin, P.; Hollebecque, A.; Arnalsteen, L.; Buob, D.; Leteurtre, E.; Caiazzo, R.; Pigeyre, M.; Verkindt, H.; Dharancy, S.; Louvet, A.; et al. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology 2009, 137, 532–540. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R.; Guidelines, R.O.T.A.O.C.P. American Association of Clinical Endocrinologists and American College of Endocrinology Comprehensive Clinical Practice Guidelines for Medical Care of Patients with Obesity. Endocr. Pract. 2016, 22, 842–884. Available online: https://www.aace.com/publications/guidelines (accessed on 10 May 2023). [CrossRef] [PubMed]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T. Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults with Overweight or Obesity without Diabetes: The STEP 8 Randomized Clinical Trial. JAMA 2022, 327, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Petit, J.-M.; Cercueil, J.-P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Vergès, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients with Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef]
- Bi, Y.; Zhang, B.; Xu, W.; Yang, H.; Feng, W.; Li, C.; Tong, G.; Li, M.; Wang, X.; Shen, S.; et al. Effects of exenatide, insulin, and pioglitazone on liver fat content and body fat distributions in drug-naive subjects with type 2 diabetes. Acta Diabetol. 2014, 51, 865–873. [Google Scholar] [CrossRef]
- Khoo, J.; Hsiang, J.; Taneja, R.; Law, N.M.; Ang, T.L. Comparative effects of liraglutide 3 mg vs structured lifestyle modification on body weight, liver fat and liver function in obese patients with non-alcoholic fatty liver disease: A pilot randomized trial. Diabetes Obes. Metab. 2017, 19, 1814–1817. [Google Scholar] [CrossRef]
- Luo, Q.; Wei, R.; Cai, Y.; Zhao, Q.; Liu, Y.; Liu, W.J. Efficacy of Off-Label Therapy for Non-alcoholic Fatty Liver Disease in Improving Non-invasive and Invasive Biomarkers: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. Front. Med. 2022, 9, 793203. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Derdemezis, C.S.; Gazi, I.F.; Nakou, E.S.; Mikhailidis, D.P.; Elisaf, M.S. Orlistat-associated adverse effects and drug interactions: A critical review. Drug Saf. 2008, 31, 53–65. [Google Scholar] [CrossRef]
- Zelber–Sagi, S.; Kessler, A.; Brazowsky, E.; Webb, M.; Lurie, Y.; Santo, M.; Leshno, M.; Blendis, L.; Halpern, Z.; Oren, R. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2006, 4, 639–644. [Google Scholar] [CrossRef]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar] [CrossRef]
- Assy, N.; Hussein, O.; Abassi, Z. Weight loss induced by orlistat reverses fatty infiltration and improves hepatic fibrosis in obese patients with non-alcoholic steatohepatitis. Gut 2007, 56, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Fincke, C.; Helinski, D.; Torgerson, S.; Hayashi, P. A pilot study of orlistat treatment in obese, non-alcoholic steatohepatitis patients. Aliment. Pharmacol. Ther. 2004, 20, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, P.M.; Harrat, Z.; Hamrioui, B.; Thellier, M.; Datry, A.; Danis, M. A simple media for isolation and culture of leishmania. Bull. Soc. Pathol. Exot. 1996, 89, 276–277. [Google Scholar] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.; Tangen, C.M.; Lucia, M.S.; Goodman, P.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; Karp, D.D.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef]
- Miller, E.R., III; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-analysis: Pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2012, 35, 66–75. [Google Scholar] [CrossRef]
- Yau, H.; Rivera, K.; Lomonaco, R.; Cusi, K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr. Diab Rep. 2013, 13, 329–341. [Google Scholar] [CrossRef]
- Bril, F.; Cusi, K. Management of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes: A Call to Action. Diabetes Care 2017, 40, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Sinha, R.A. Current treatment paradigms emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 Analogs Liraglutide and Semaglutide Reduce Atherosclerosis in ApoE(-/-) and LDLr(-/-) Mice by a Mechanism That Includes Inflammatory Pathways. JACC Basic. Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef]
- Ho, P.P.; Steinman, L. Obeticholic acid, a synthetic bile acid agonist of the farnesoid X receptor, attenuates experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2016, 113, 1600–1605. [Google Scholar] [CrossRef]
- Guirguis, E.; Grace, Y.; Bolson, A.; DellaVecchia, M.J.; Ruble, M. Emerging therapies for the treatment of nonalcoholic steatohepatitis: A systematic review. Pharmacotherapy 2021, 41, 315–328. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 2019, 84, 105803. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) alpha/gamma/delta Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]
- Wettstein, G.; Luccarini, J.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrêne, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Hodson, L. BA Fielding, Stearoyl-CoA desaturase: Rogue or innocent bystander? Prog. Lipid. Res. 2013, 52, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Walle, P.; Takkunen, M.; Männistö, V.; Vaittinen, M.; Lankinen, M.; Kärjä, V.; Käkelä, P.; Ågren, J.; Tiainen, M.; Schwab, U.; et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Issandou, M.; Bouillot, A.; Brusq, J.M.; Forest, M.C.; Grillot, D.; Guillard, R.; Martin, S.; Michiels, C.; Sulpice, T.; Daugan, A. Pharmacological inhibition of stearoyl-CoA desaturase 1 improves insulin sensitivity in insulin-resistant rat models. Eur. J. Pharmacol. 2009, 618, 28–36. [Google Scholar] [CrossRef]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernández-Ramos, D.; Martínez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; Delacruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Hershkovitz, A.; Rosenthal-Galili, Z.; Zuckerman, E.; et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Rubin, V.R.; Bojanic, K.; Smolic, M.; Rubin, J.; Tabll, A.; Smolic, R. An Update on Efficacy and Safety of Emerging Hepatic Antifibrotic Agents. J. Clin. Transl. Hepatol. 2021, 9, 60–70. [Google Scholar]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Reciprocal Crosstalk between Autophagic and Endocrine Signaling in Metabolic Homeostasis. Endocr. Rev. 2017, 38, 69–102. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; You, S.-H.; Zhou, J.; Siddique, M.M.; Bay, B.-H.; Zhu, X.; Privalsky, M.L.; Cheng, S.-Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Sherman, S.I.; Ringel, M.D.; Smith, M.J.; Kopelen, H.A.; Zoghbi, W.A.; Ladenson, P.W. Augmented hepatic and skeletal thyromimetic effects of tiratricol in comparison with levothyroxine. J. Clin. Endocrinol. Metab. 1997, 82, 2153–2158. [Google Scholar] [CrossRef]
- Kelly, M.J.; Pietranico-Cole, S.; Larigan, J.D.; Haynes, N.E.; Reynolds, C.H.; Scott, N.; Vermeulen, J.; Dvorozniak, M.; Conde-Knape, K.; Huang, K.-S.; et al. Discovery of 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dio xo-2,3,4,5-tetrahydro [1,2,4]triazine-6-carbonitrile (MGL-3196), a Highly Selective Thyroid Hormone Receptor beta agonist in clinical trials for the treatment of dyslipidemia. J. Med. Chem. 2014, 57, 3912–3923. [Google Scholar]
- Taub, R.; Chiang, E.; Chabot-Blanchet, M.; Kelly, M.J.; Reeves, R.A.; Guertin, M.-C.; Tardif, J.-C. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted thyroid hormone receptor-beta agonist. Atherosclerosis 2013, 230, 373–380. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Frias, J.P.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients with NASH. Hepatol. Commun. 2021, 5, 573–588. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameters | NCEP ATP3 2005 |
|---|---|
| Number of abnormalities | ≥3 of: |
| Glucose | Fasting glucose ≥ 5.6 mmol/L (100 mg/dL) or drug treatment for elevated blood glucose |
| HDL cholesterol | <1.0 mmol/L (40 mg/dL) (men); <1.3 mmol/L (50 mg/dL) (women) or drug treatment for low HDL cholesterol § |
| Triglycerides | ≥1.7 mmol/L (150 mg/dL) or drug treatment for elevated triglycerides § |
| Obesity | Waist ≥ 102 cm (men) or ≥88 cm (women) * |
| Hypertension | ≥130/85 mmHg or drug treatment for hypertension |
| Theorized Mechanism of Gut Microbiome Alteration | Specific Gut Microbiome Alteration | Contribution to Liver Disease Progression |
|---|---|---|
| Increased production and absorption of gut short-chain fatty acids (SCFA) | Increased abundance of Firmicutes and reduced abundance of Bacteroidetes [35,36] | Increased SCFA leading to increased obesity [37,38] |
| Altered dietary choline metabolism | Reduced Gammaproteobacteria abundance and Increased Erysipelotrichi abundance [39,40] | Reduced choline caused hepatic steatosis [41] Altered microbiota convert dietary choline to hepatotoxic and inflammatory methylamine [41] |
| Reduced bile acid production | Unknown | Reduced activation of farnesoid X receptor and TGR5 by bile acids, leading to reduced inhibition of pro-inflammatory cytokine production and increased hepatic expression of TNFα and IL-18 [42]. |
| Gut permeability alterations and release of endotoxin | Increased proportion of gram-negative bacteria [43] | Increased production of lipopolysaccharide which is an endotoxin, leading to increased delivery to the liver and resultant steatohepatitis [44]. |
| Increased dietary Saturated Fat intake | Reduced microbial diversity and increased Firmicutes-to Bacteroidetes ratio [45] | Increased SCFA production leading to increased obesity [37,38] |
| Increased dietary fructose intake | Decreased Bifidobacterium and Lactobacillus abundance [46] | Increased endotoxinemia leading to steatohepatitis [46,47] |
| Comorbid chronic kidney disease | Increased abundance of urea-metabolizing microbial strains at the expense of carbohydrate-fermenting strains [48,49] | Accumulation of uremic toxic metabolites (URMs) which disrupts intestinal tight junctions and facilitates passage of lipopolysaccharides causing steatohepatitis [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beiriger, J.; Chauhan, K.; Khan, A.; Shahzad, T.; Parra, N.S.; Zhang, P.; Chen, S.; Nguyen, A.; Yan, B.; Bruckbauer, J.; et al. Advancements in Understanding and Treating NAFLD: A Comprehensive Review of Metabolic-Associated Fatty Liver Disease and Emerging Therapies. Livers 2023, 3, 637-656. https://doi.org/10.3390/livers3040042
Beiriger J, Chauhan K, Khan A, Shahzad T, Parra NS, Zhang P, Chen S, Nguyen A, Yan B, Bruckbauer J, et al. Advancements in Understanding and Treating NAFLD: A Comprehensive Review of Metabolic-Associated Fatty Liver Disease and Emerging Therapies. Livers. 2023; 3(4):637-656. https://doi.org/10.3390/livers3040042
Chicago/Turabian StyleBeiriger, Jacob, Kashyap Chauhan, Adnan Khan, Taha Shahzad, Natalia Salinas Parra, Peter Zhang, Sarah Chen, Anh Nguyen, Brian Yan, John Bruckbauer, and et al. 2023. "Advancements in Understanding and Treating NAFLD: A Comprehensive Review of Metabolic-Associated Fatty Liver Disease and Emerging Therapies" Livers 3, no. 4: 637-656. https://doi.org/10.3390/livers3040042
APA StyleBeiriger, J., Chauhan, K., Khan, A., Shahzad, T., Parra, N. S., Zhang, P., Chen, S., Nguyen, A., Yan, B., Bruckbauer, J., & Halegoua-DeMarzio, D. (2023). Advancements in Understanding and Treating NAFLD: A Comprehensive Review of Metabolic-Associated Fatty Liver Disease and Emerging Therapies. Livers, 3(4), 637-656. https://doi.org/10.3390/livers3040042