
In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results †



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
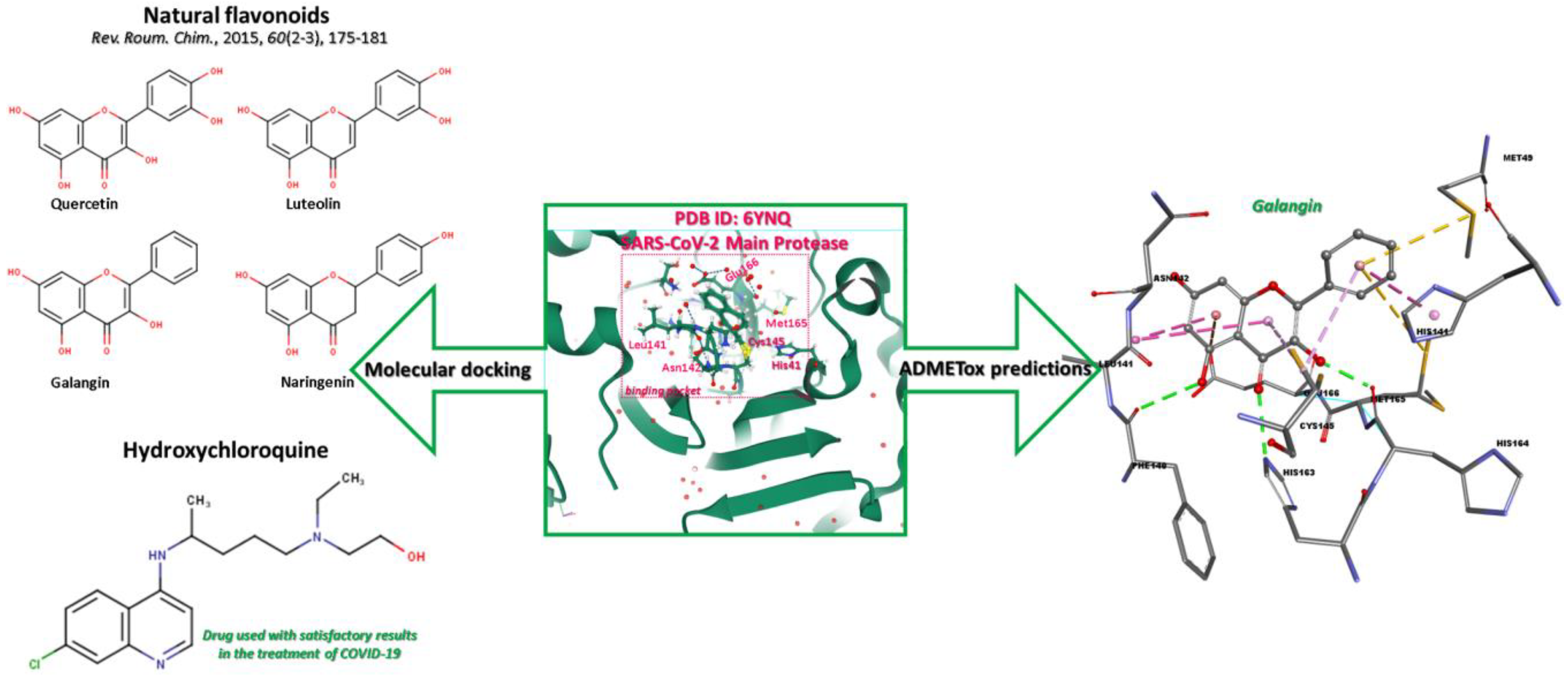
2.1. Workflow
2.2. Ligands Preparation
2.3. Protein Preparation
2.4. Molecular Docking
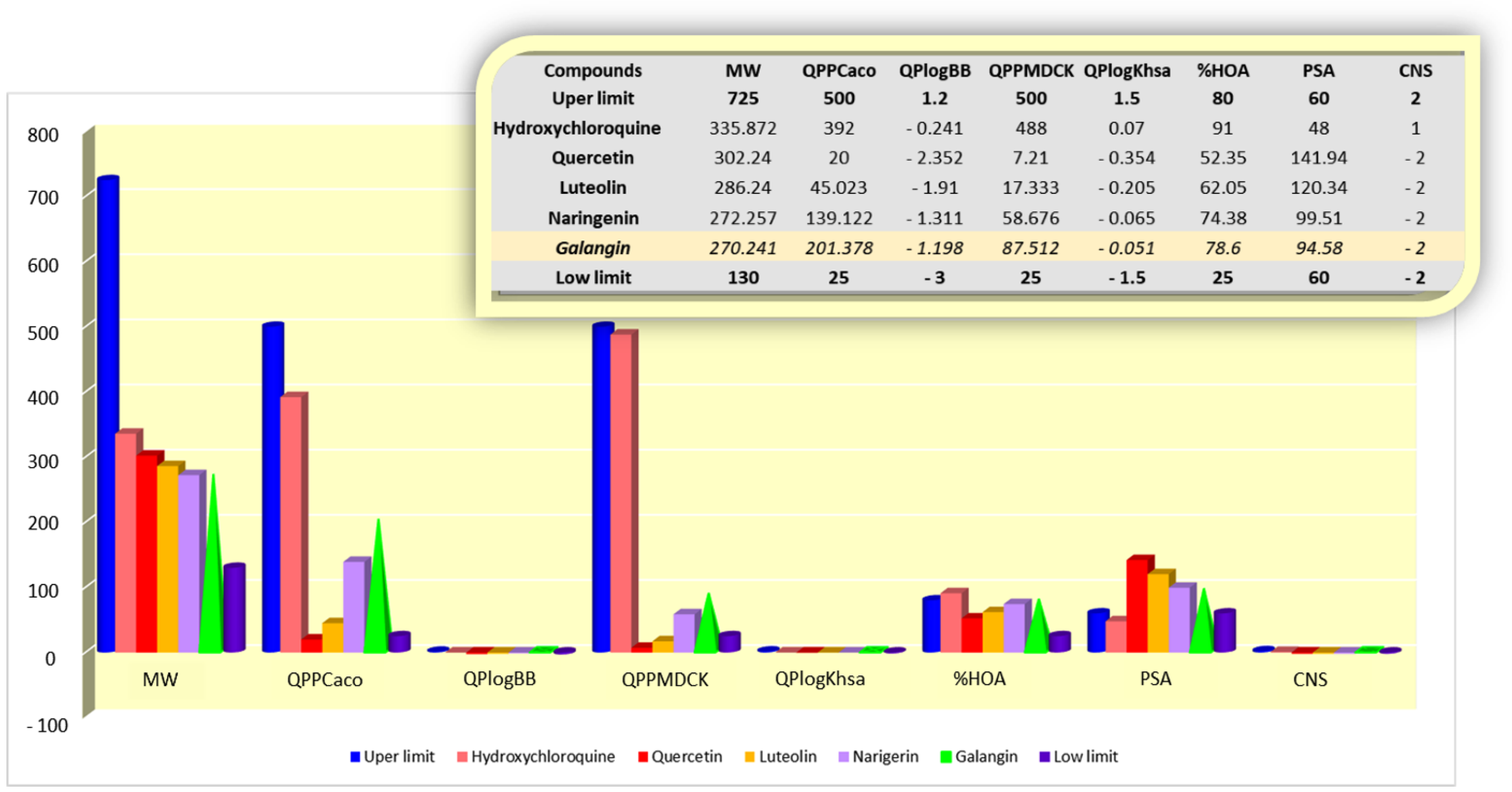
2.5. Prediction of Pharmacokinetic Profile
- QPPCaco—predicted apparent Caco-2 cell permeability in nm/s: >500 (great) and <25 (poor);
- QPPMDCK—predicted apparent MDCK cell permeability in nm/s: >500 (great) and <25 (poor);
- QPlogKhsa—prediction of binding to human serum albumin: −1.5 (low) to 1.5 (high);
- QlogBB—predicted brain/blood partition coefficient: −3.0 (low) to 1.2 (easy permeability);
- CNS—predicted central nervous system activity:–2 (low permeability) to +2 (high permeability;
- PSA—van der Waals surface area of polar nitrogen and oxygen atoms: >60 does not cross the blood–brain barrier and <60 to cross the blood–brain barrier;
- % HOA—predicted human oral absorption on 0 to 100% scale: >80% (high), 25–80% (medium) and <25% (poor).
3. Results and Discussions
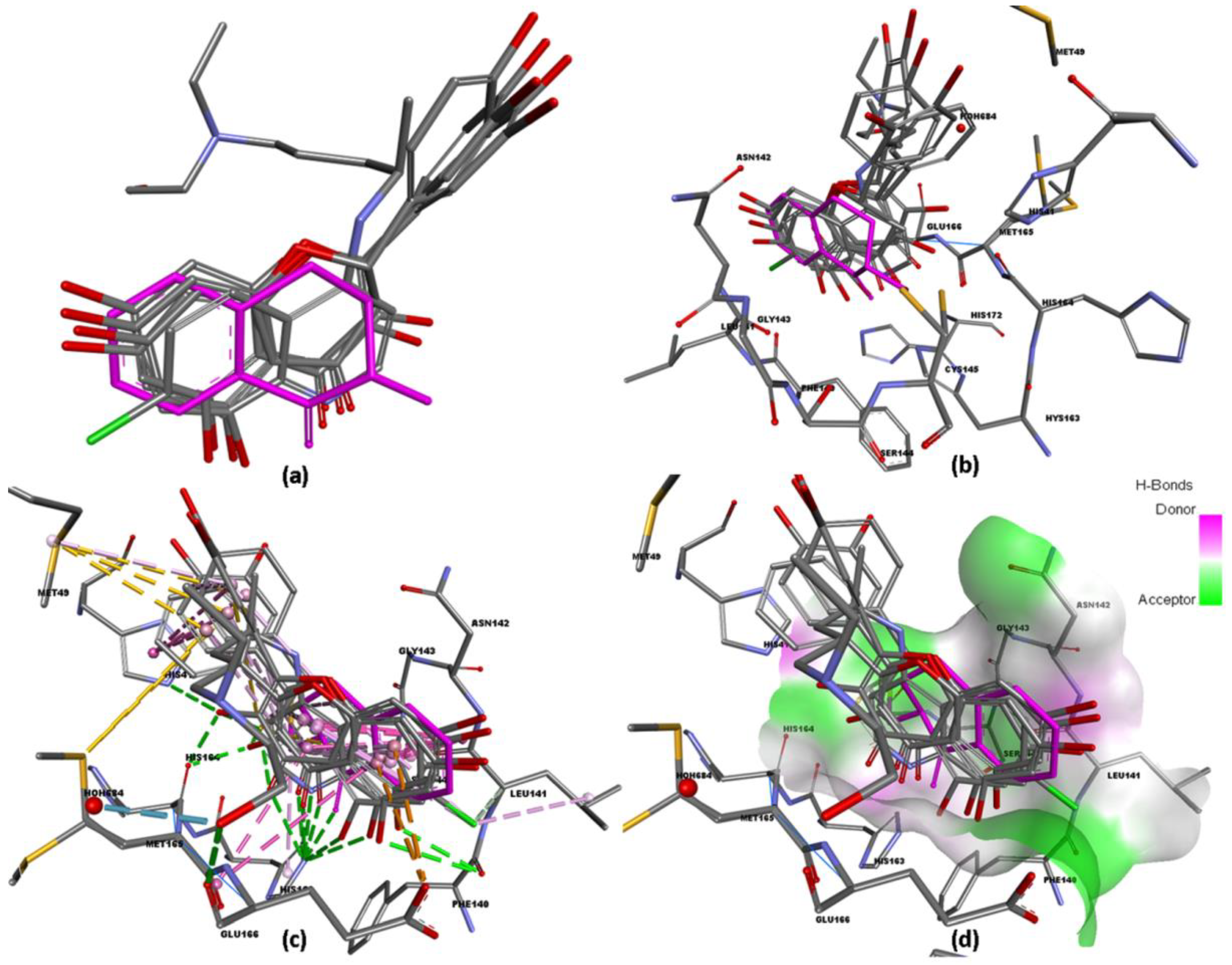
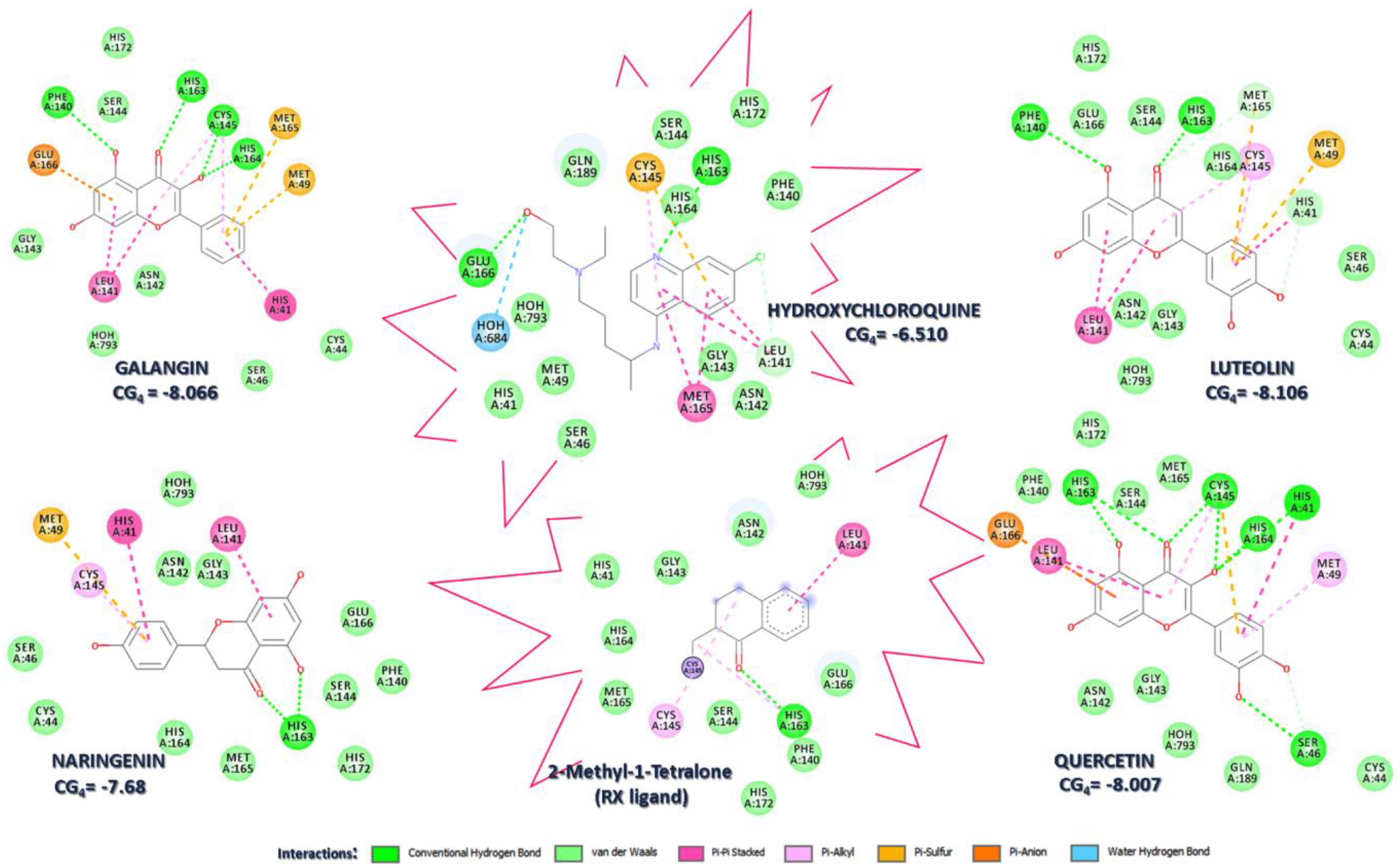
3.1. Molecular Docking Analysis
3.2. Pharmacokinetic and Toxicological Properties Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acter, T.; Uddin, N.; Das, J.; Akhter, A.; Choudhury, T.R.; Kim, S. Evolution of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-Cov-2) as Coronavirus Disease 2019 (COVID-19) pandemic: A global health emergency. Sci. Total Environ. 2020, 730, 138996. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Treatment for COVID-19. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 6 October 2020).
- Guarner, J. Three emerging coronaviruses in two decades: The story of SARS, MERS, and now COVID-19. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Hage-Melim, L.I.D.S.; Federico, L.B.; de Oliveira, N.K.S.; Francisco, V.C.C.; Correia, L.C.; de Lima, H.B.; Gomes, S.Q.; Barcelos, M.P.; Francischini, I.A.G.; da Silva, C.H.T.P. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020, 256, 117963. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of natural products as potential inhibitors of COVID-19 main protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef]
- Pandey, A.K.; Verma, S. An in-silico evaluation of dietary components for structural inhibition of SARS-Cov-2 main protease. J. Biomol. Struct. Dyn. 2020, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.-E.; Lowe, H.; Toyang, N. Potential of flavonoid-inspired phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef]
- Russo, M.; Moccia, S.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Roles of flavonoids against coronavirus infection. Chem. Biol. Interact. 2020, 328, 109211. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- D’Arminio Monforte, A.; Tavelli, A.; Bai, F.; Marchetti, G.; Cozzi-Lepri, A. Effectiveness of hydroxychloroquine in COVID-19 disease: A done and dusted deal? Int. J. Infect. Dis. 2020, 99, 75–76. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge structural database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Bora, A.; Avram, S.; Crisan, L.; Halip, L.; Curpan, R.; Pacureanu, L.; Ciucanu, I. Theoretical investigations of quercetin and its analogues as anticancer agents. Rev. Roum. Chim. 2015, 60, 175–181. [Google Scholar]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. QikProp 4.4; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020, 57, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Arshad, S.; Kilgore, P.; Chaudhry, Z.S.; Jacobsen, G.; Wang, D.D.; Huitsing, K.; Brar, I.; Alangaden, G.J.; Ramesh, M.S.; McKinnon, J.E.; et al. Henry Ford COVID-19 Task Force. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalised with COVID-19. Int. J. Infect. Dis. 2020, 97, 396–403. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bora, A.; Pacureanu, L.; Crisan, L. In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results. Chem. Proc. 2021, 3, 25. https://doi.org/10.3390/ecsoc-24-08343
Bora A, Pacureanu L, Crisan L. In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results. Chemistry Proceedings. 2021; 3(1):25. https://doi.org/10.3390/ecsoc-24-08343
Chicago/Turabian StyleBora, Alina, Liliana Pacureanu, and Luminita Crisan. 2021. "In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results" Chemistry Proceedings 3, no. 1: 25. https://doi.org/10.3390/ecsoc-24-08343
APA StyleBora, A., Pacureanu, L., & Crisan, L. (2021). In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results. Chemistry Proceedings, 3(1), 25. https://doi.org/10.3390/ecsoc-24-08343