
Molecular Docking Studies on Synthetic Therapeutic Agents for COVID-19 †



Abstract
:1. Introduction
2. Molecular Docking Studies
2.1. SARS-CoV-2 Main Protease
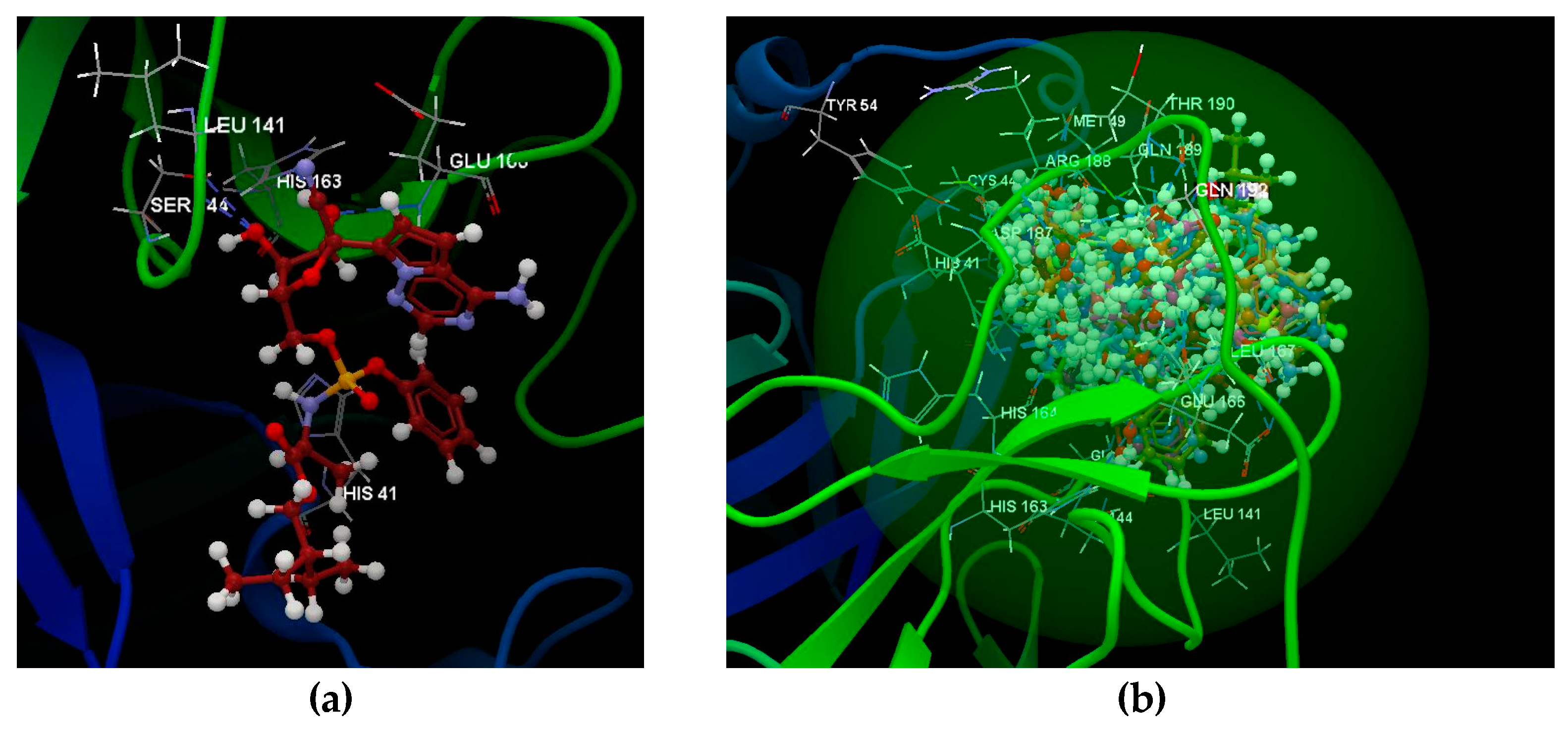
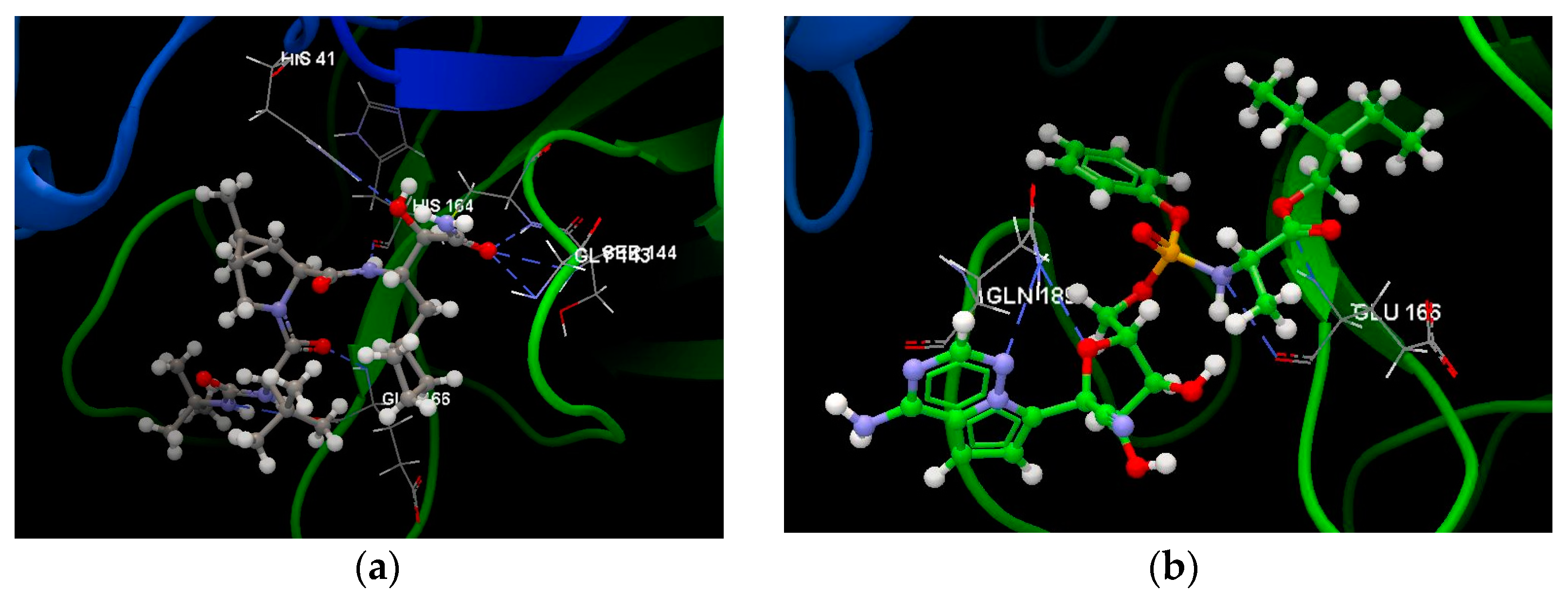
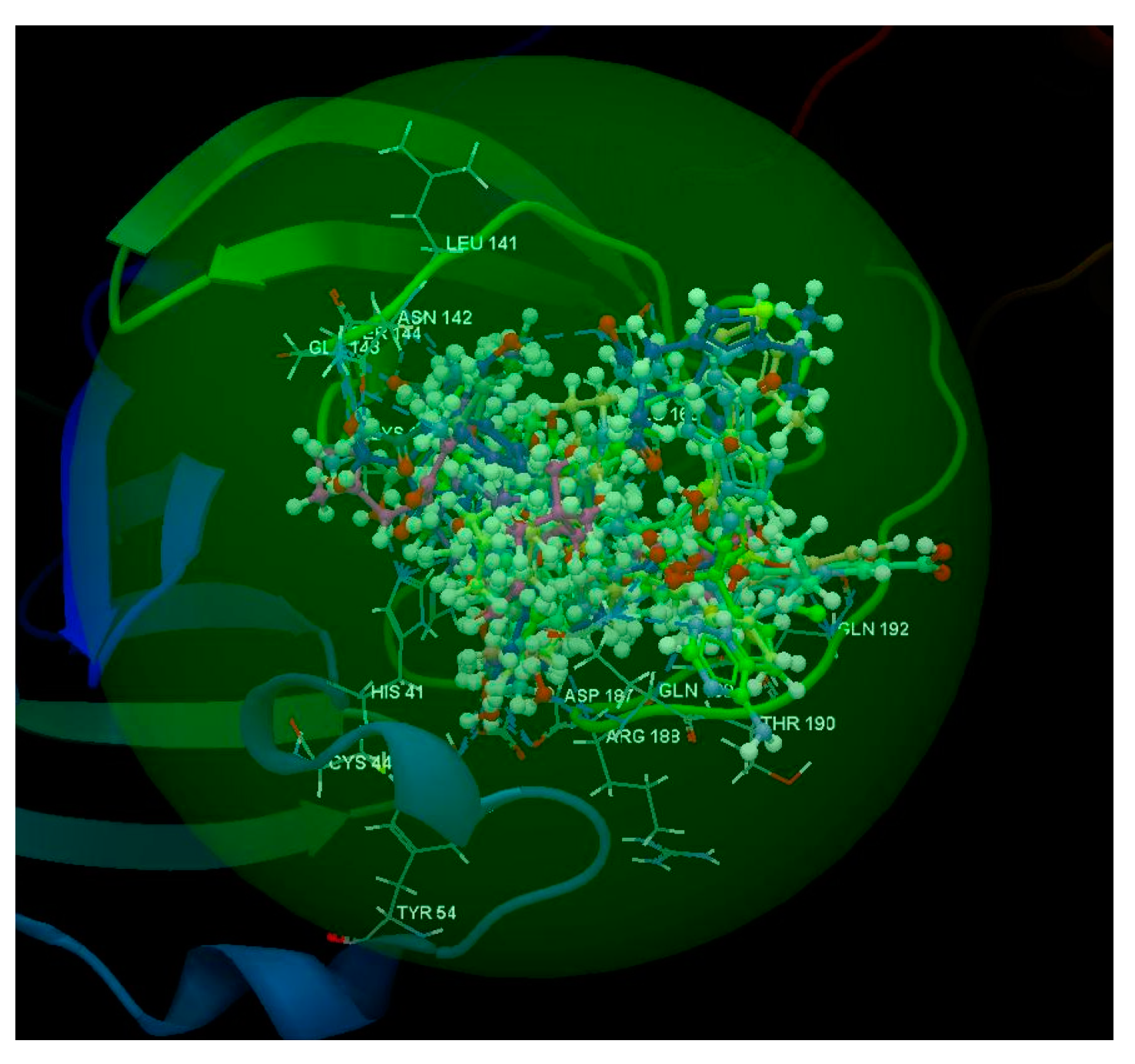
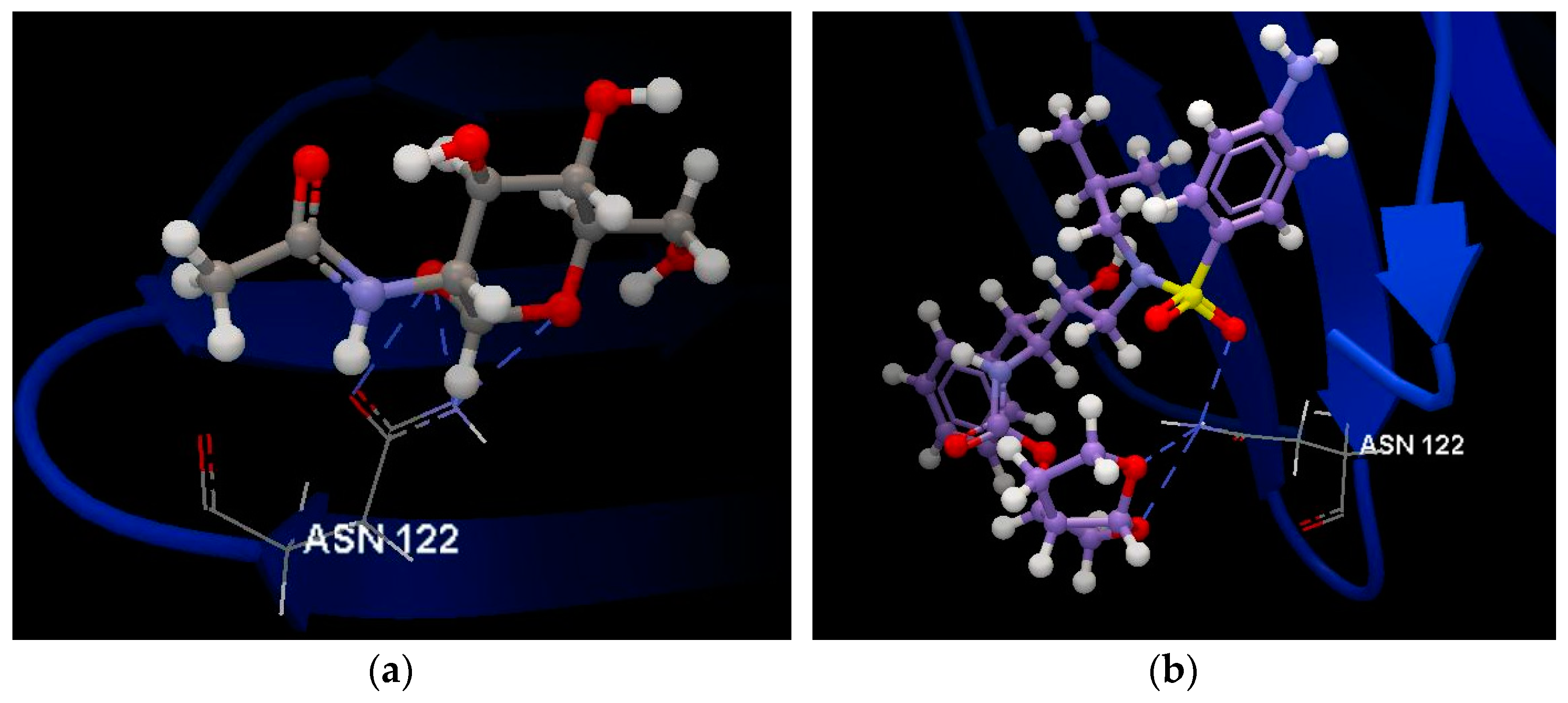
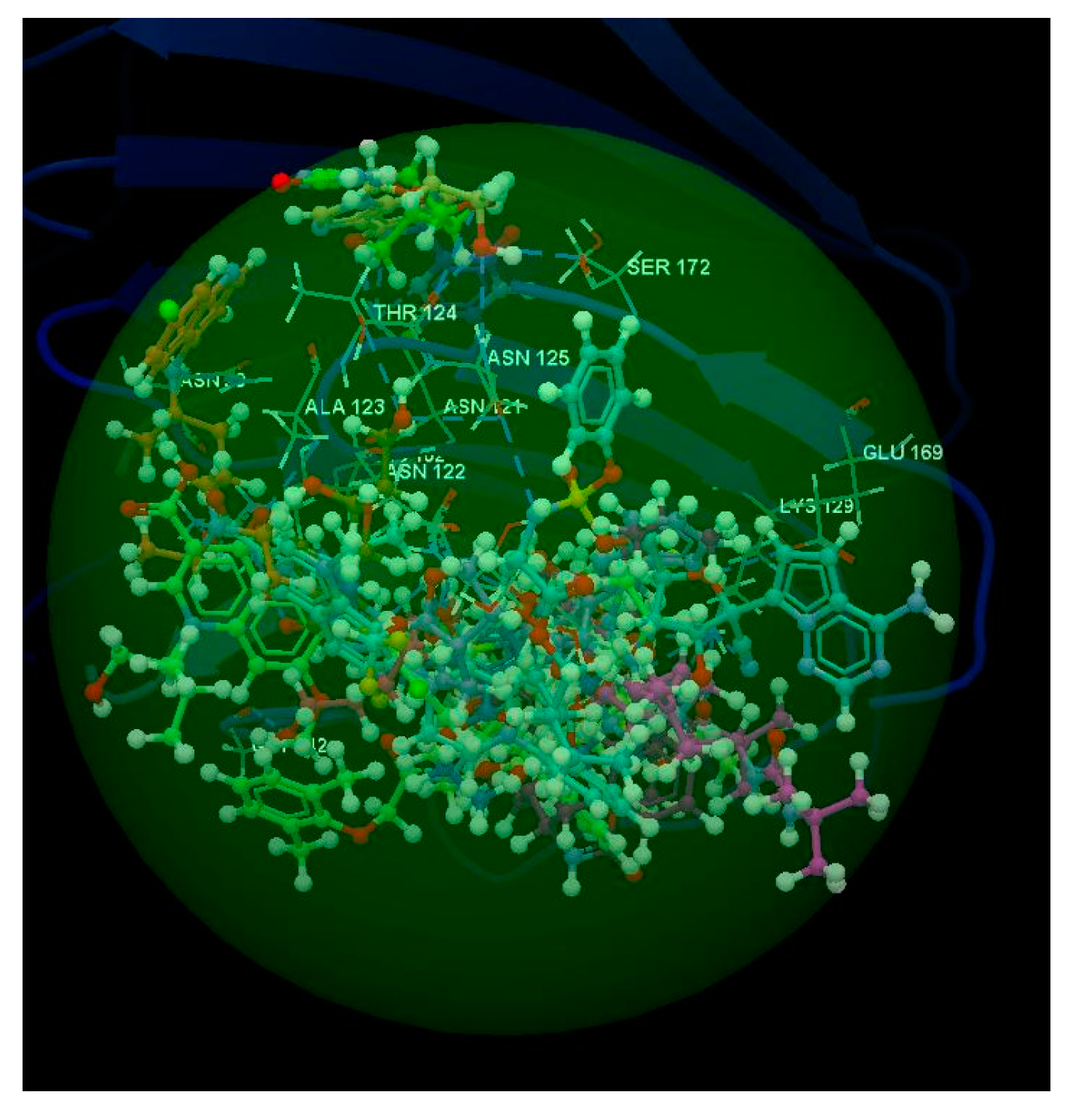
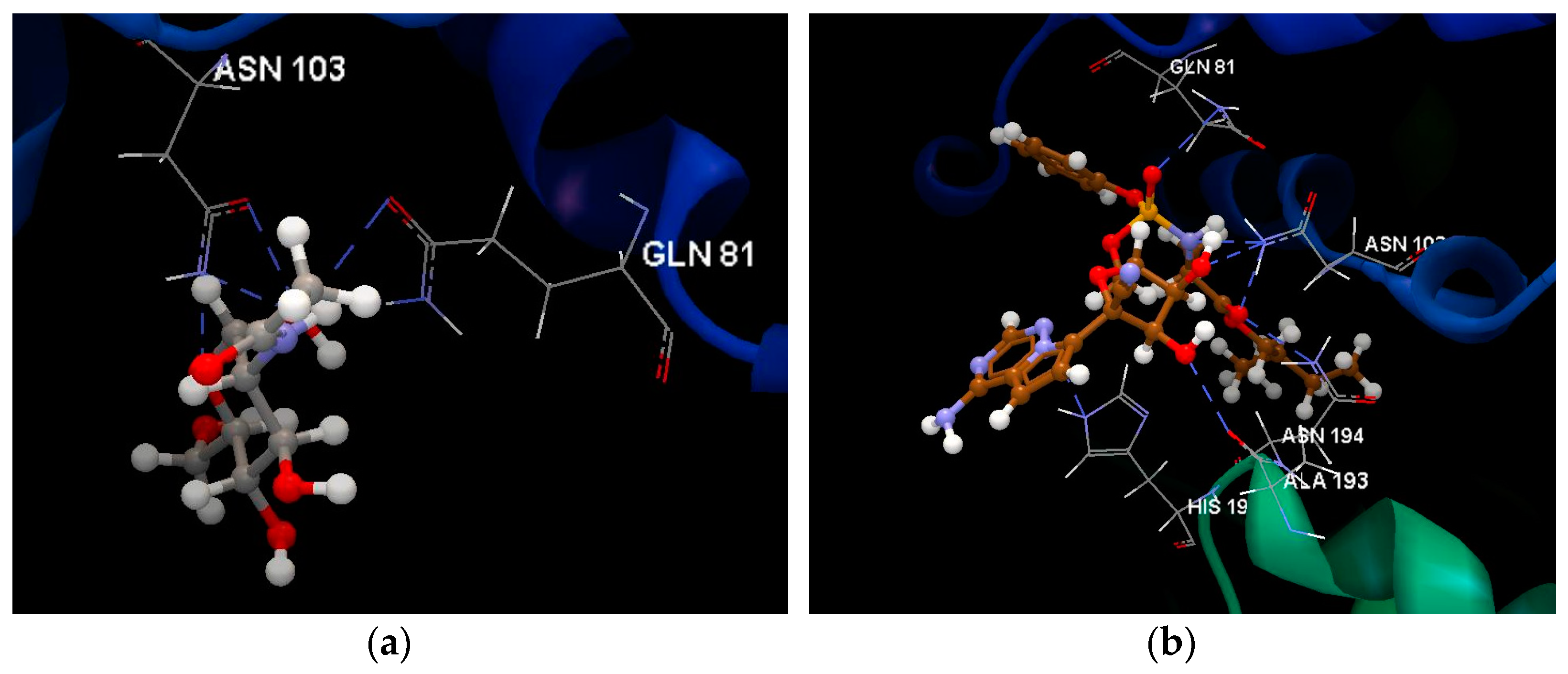
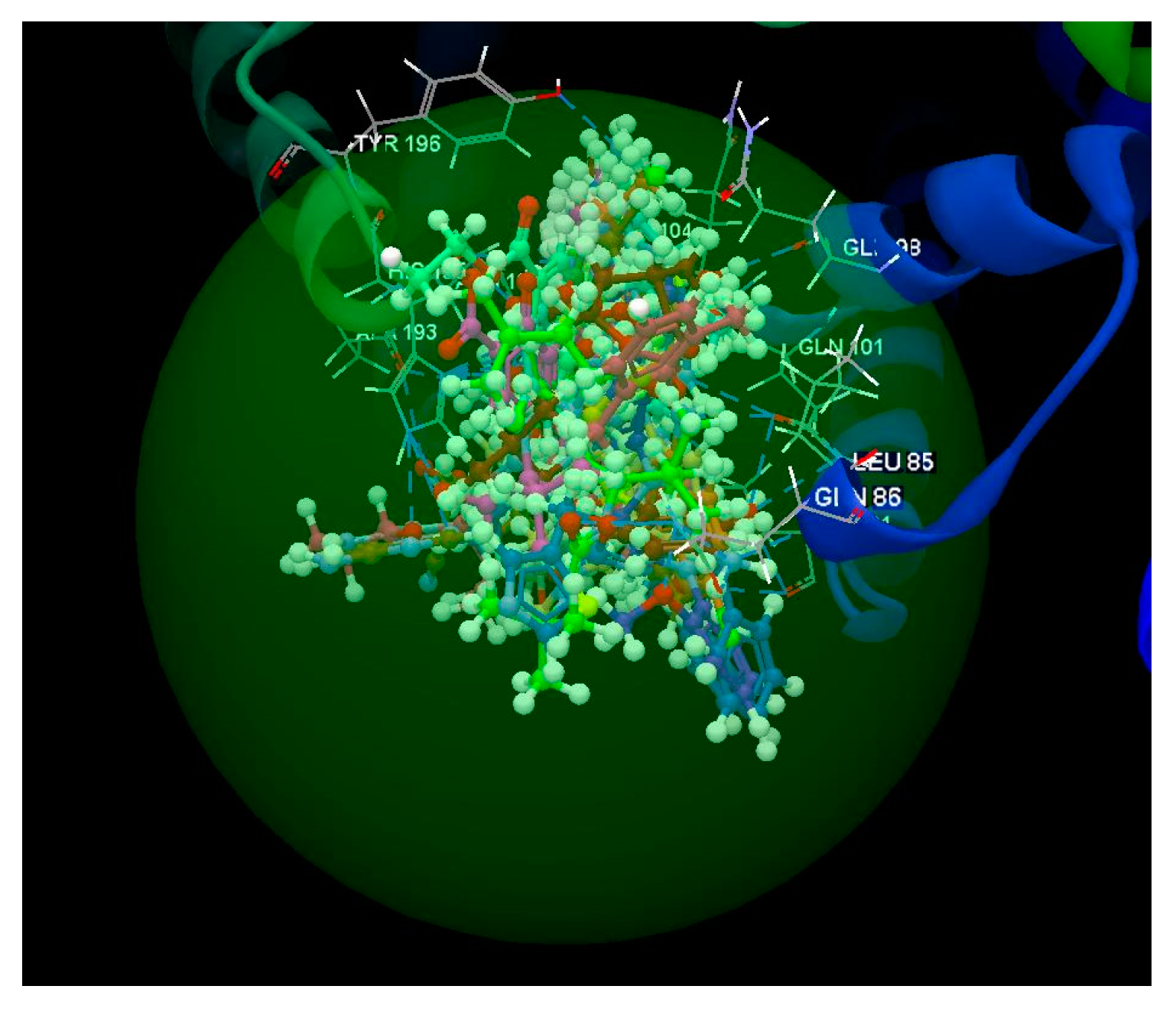
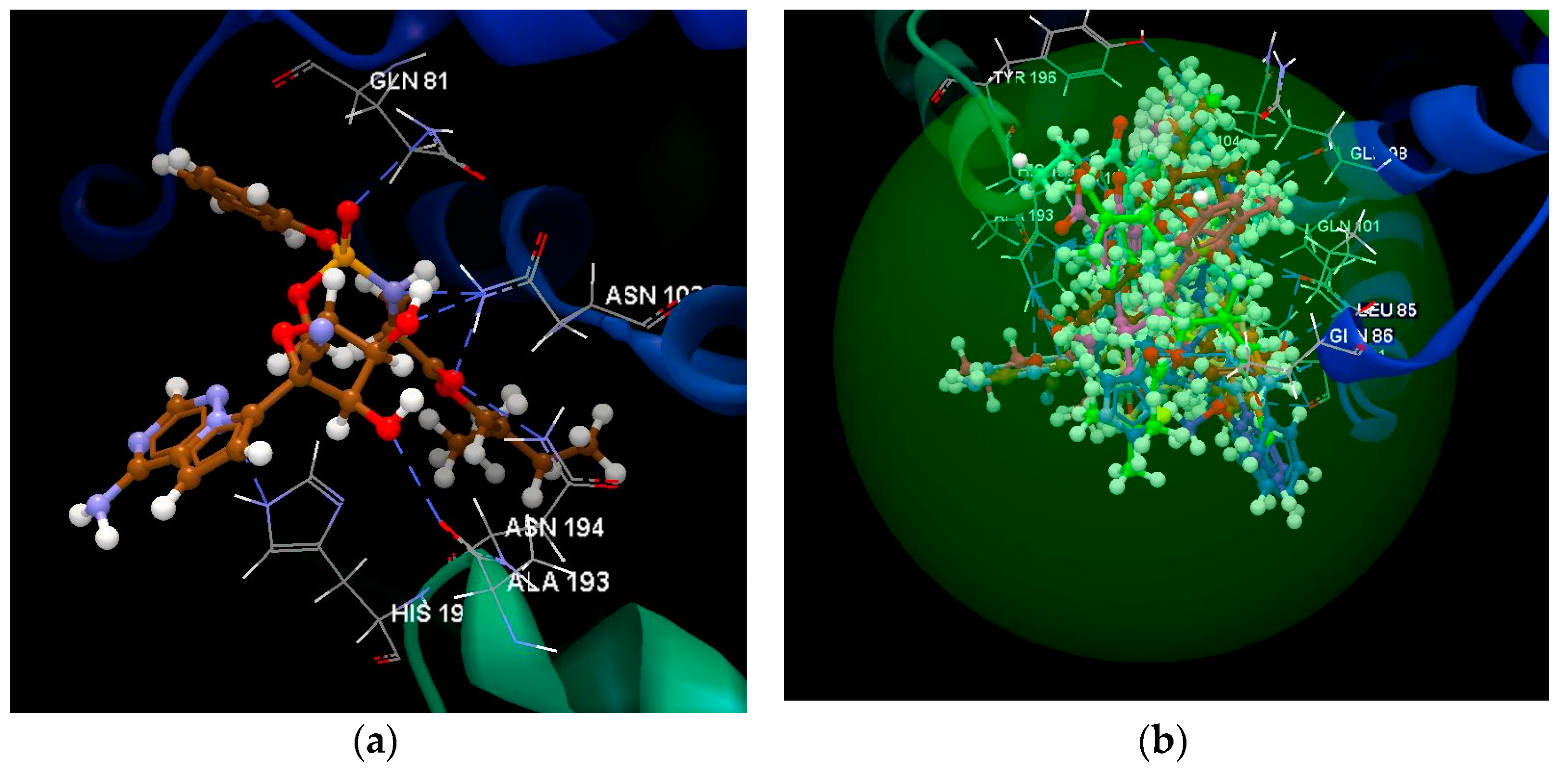
2.1.1. SARS-CoV-2 Main Protease Receptor PD ID: 6W63
2.1.2. SARS-CoV-2 Main Protease Receptor PD ID: 6WNP
2.2. SARS-CoV-2 Spike Glycoprotein
2.3. SARS-CoV-2 Chimeric Receptor-Binding Domain Complexed with Its Receptor Human ACE2
2.4. SARS-CoV-2 RNA-Dependent RNA Polymerase
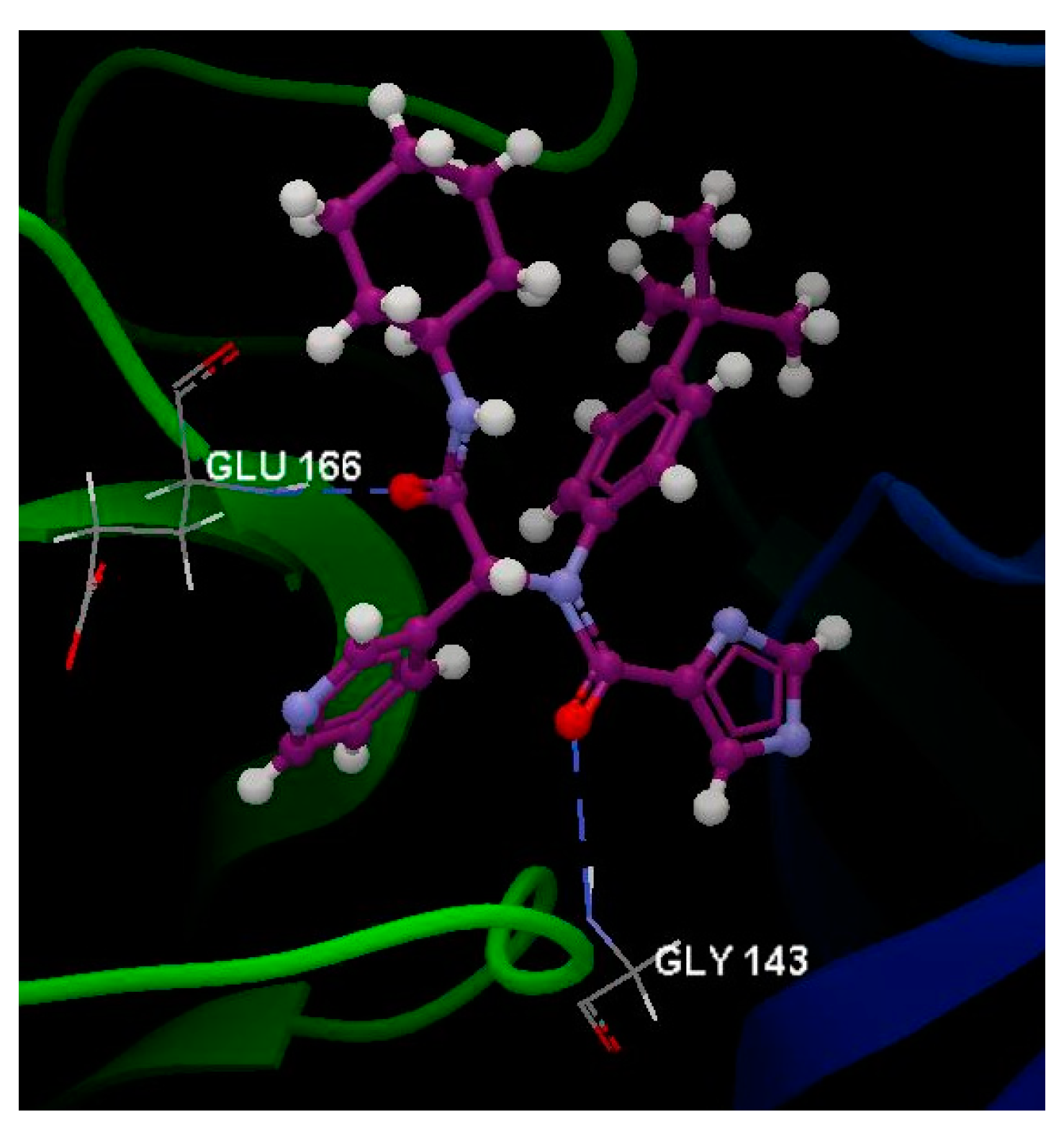
2.5. SARS-CoV-2 3CL Protease (3CL pro)
3. Conclusions
- protease inhibitors: Lopinavir, Ritonavir, Darunavir
- integrase inhibitors: Elvitegravir and Remdesivir, originally developed for the treatment of Marburg virus, Ebola virus and Cueva virus infections.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohapatra, R.K.; Pintilie, L.; Kandi, V.; Sarangi, A.K.; Das, D.; Sahu, R.; Perekhoda, L. The recent challenges of highly contagious COVID-19, causing respiratory infections: Symptoms, diagnosis, transmission, possible vaccines, animal models, and immunotherapy. Chem. Biol. Drug Des. 2020. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, L.; Kuo, H.-C.D.; Shannar, A.; Peter, R.; Chou, P.J.; Li, S.; Hudlikar, R.; Liu, X.; Liu, Z.; et al. An Update on Current Therapeutic Drugs Treating COVID-19. Curr. Pharmacol. Rep. 2020, 6, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Jha, T. Fight against novel coronavirus: A perspective of medicinal chemists. Eur. J. Med. Chem. 2020, 201, 112559. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, S.M.V.; Santos Soares, J.C.; dos Santos, K.L.; Alves, J.E.F.; Ribeiro, A.G.; Jacob, I.T.T.; da Silva Ferreira, C.J.; dos Santos, J.C.; de Oliveira, J.F.; de Carvalho Junior, L.B.; et al. COVID-19 therapy: What weapons do we bring into battle? Bioorg. Med. Chem. 2020, 28, 115757. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef] [PubMed]
- ElFiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef] [PubMed]
- ElFiky, A.A. Corrigendum to “Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARSCoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study” [Life Sci. 253 (2020) 117592]. Life Sci. 2020, 253, 118350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, W.B.; Mendanha, S.A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar] [CrossRef] [PubMed]
- CLC Drug Discovery Workbench, 2015, QIAGEN Aarhus A/S, Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark.
- Spartan’14; Wavefunction, Inc.: Irvine, CA, USA, 2013.
- Witvrouw, M.; Daelemans, D.; Pannecouque, C.; Neyts, J.; Andrei, G.; Snoeck, R.; Vandamme, A.-M.; Balzarini, J.; Desmyter, J.; Baba, M.; et al. Broad-Spectrum Antiviral Activity and Mechanism of Antiviral Action of the Fluoroquinolone Derivative K-12. Antivir. Chem. Chemother. 1998, 9, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, K.; Shi, W. Quinolone derivatives: Potential anti-HIV agent—development and application. Arch. Pharm. Chem. Life Sci. 2019, 352, 1900045. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecular 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
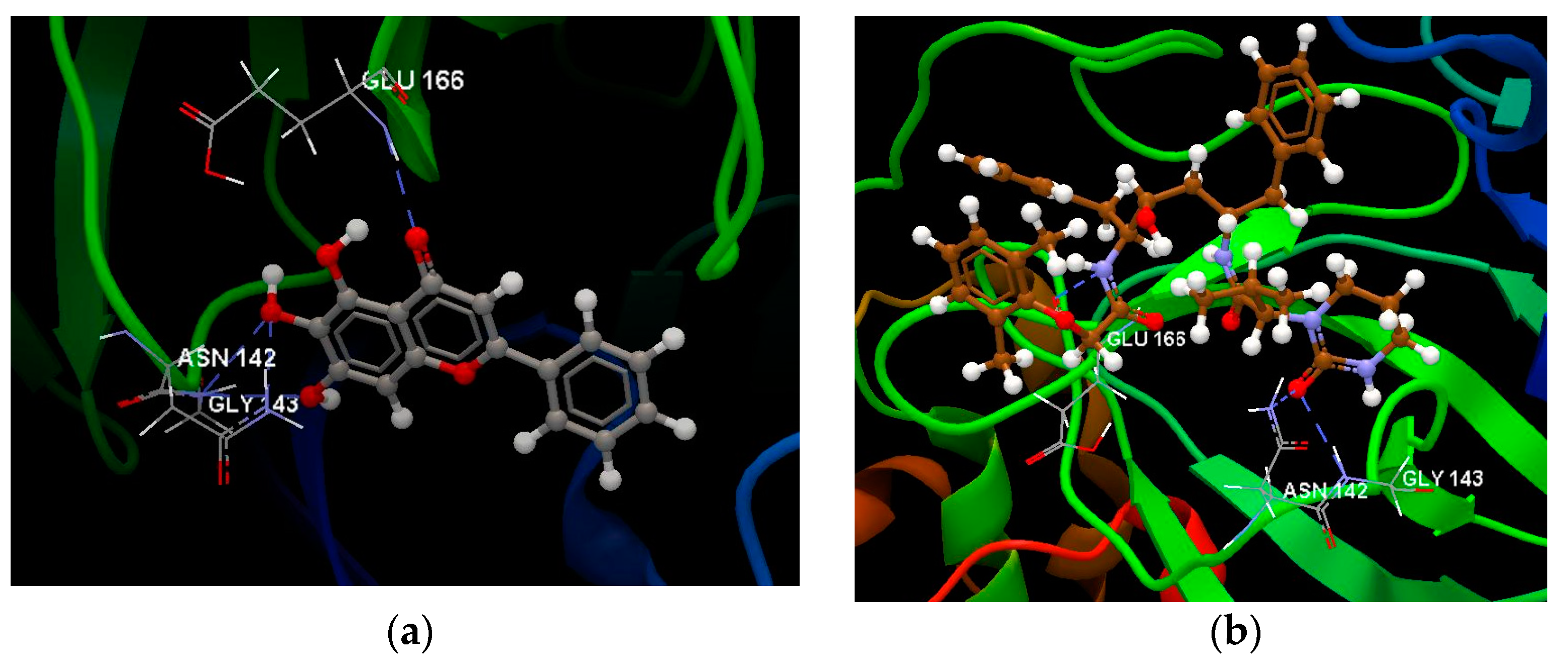{kind=link}
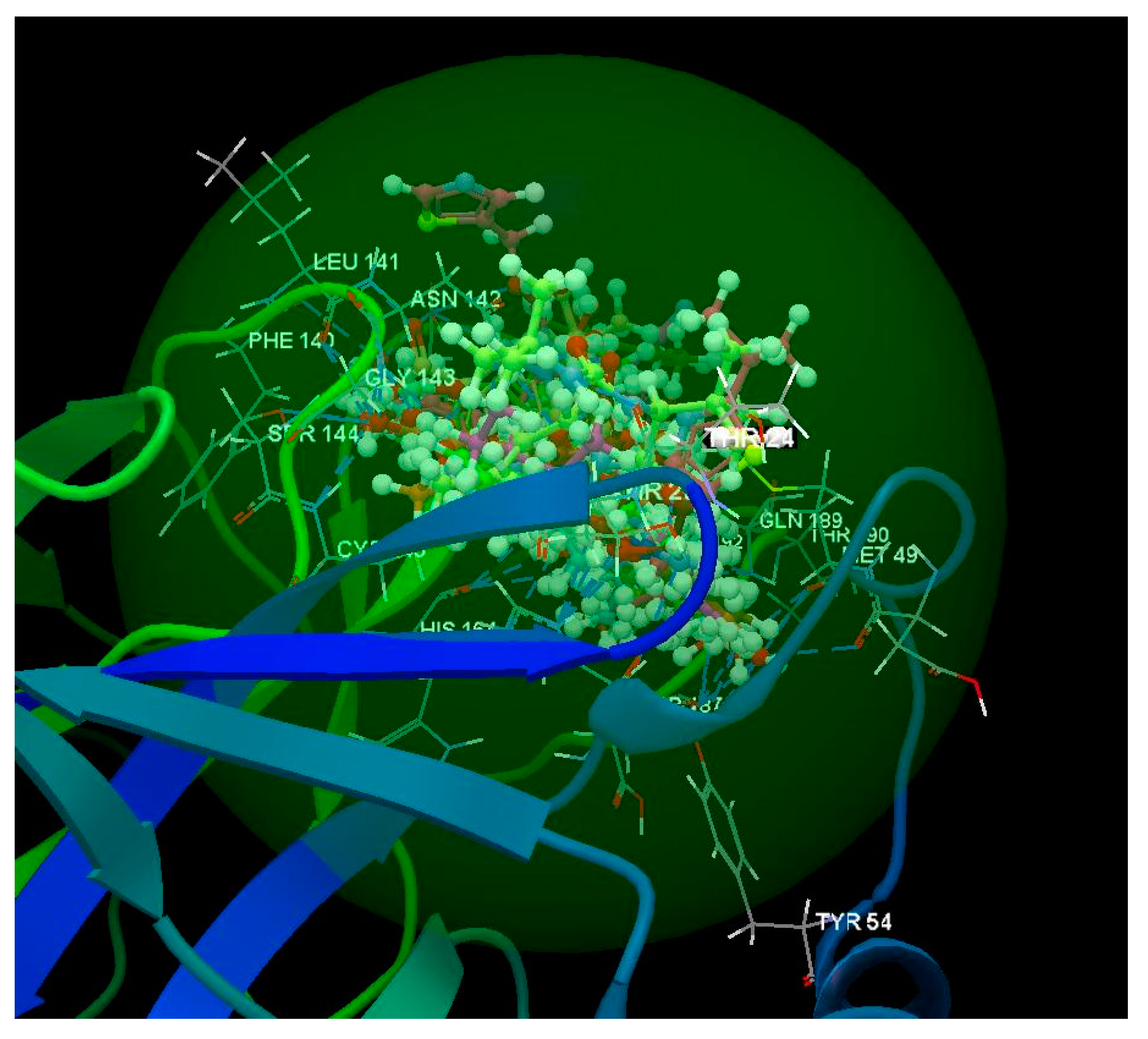{kind=link}
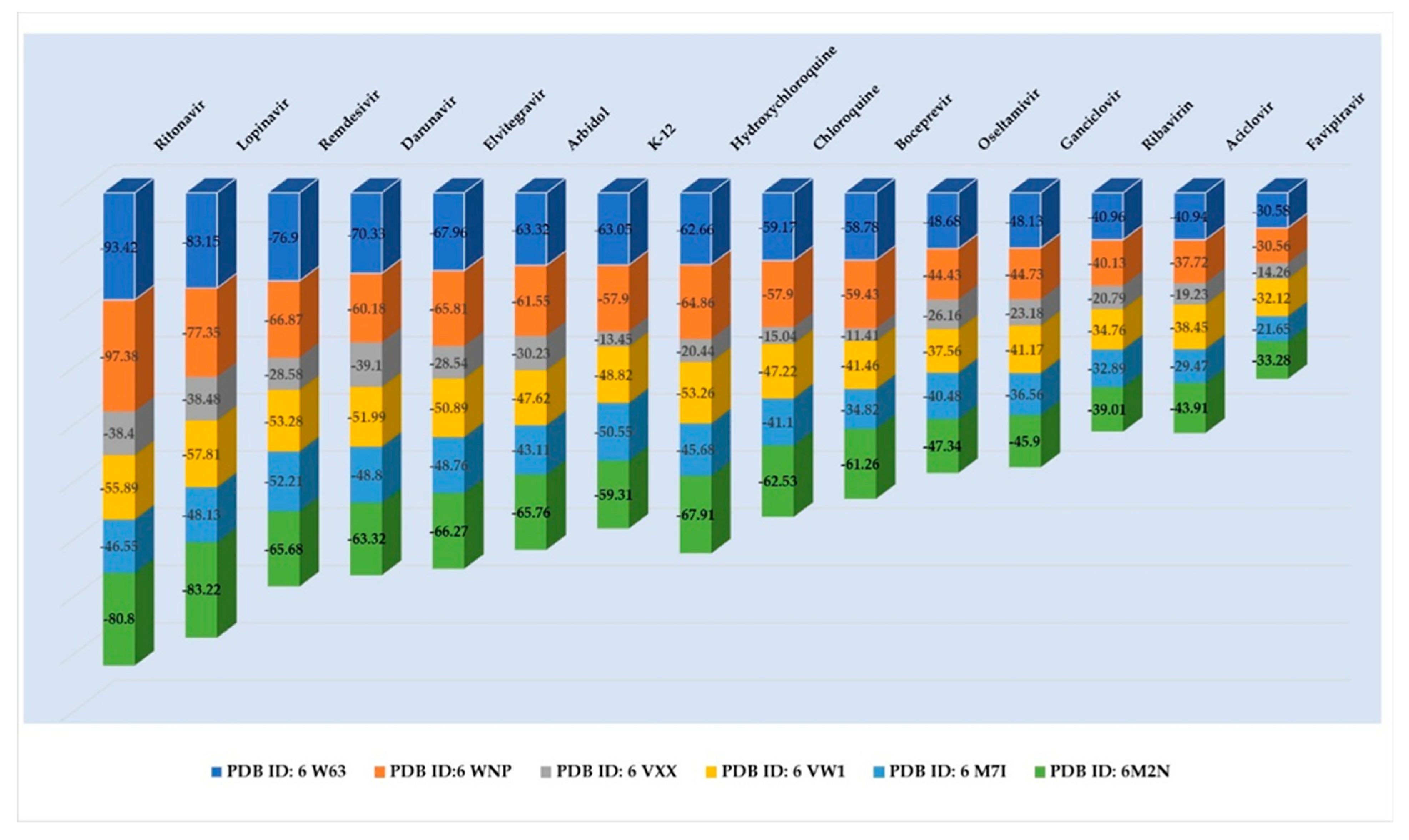
| Ligand | PDB ID: 6W63 | PDB ID: 6WNP | PDB ID: 6VXX | PDB ID: 6VW1 | PDB ID: 6M71 | PDB ID:6M2N | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | RMSD [Å] | Score | RMSD [Å] | Score | RMSD [Å] | Score | RMSD[Å] | Score | RMSD[Å] | Score | RMSD[Å] | |
| Co-crystallized | −56.65 | 0.90 | −63.95 | 0.80 | −18.42 | 0.51 | −32.77 | 0.20 | - | - | −53.49 | 0.37 |
| Ritonavir | −93.42 | 2.98 | −97.38 | 3.54 | −38.40 | 5.17 | −55.89 | 3.16 | −46.55 | 2.49 | −80.80 | 3.24 |
| Lopinavir | −83.15 | 2.15 | −77.35 | 3.48 | −38.48 | 4.69 | −57.81 | 2.67 | −48.13 | 2.35 | −83.22 | 0.98 |
| Remdesivir | −76.90 | 1.89 | −66.87 | 1.52 | −28.58 | 3.78 | −53.28 | 2.95 | −52.21 | 2.28 | 65.68 | 1.33 |
| Darunavir | −70.33 | 2.14 | −60.18 | 1.84 | −39.10 | 4.06 | −51.99 | 1.94 | −48.80 | 1.34 | −63.32 | 1.94 |
| Elvitegravir | −67.96 | 0.23 | −65.81 | 0.58 | −28.54 | 2.10 | −50.89 | 0.40 | −48.76 | 0.06 | −66.27 | 0.07 |
| Arbidol | −63.32 | 0.23 | −61.55 | 0.48 | −30.23 | 1.38 | −47.62 | 0.87 | −43.11 | 0.49 | −65.76 | 0.04 |
| K-12 | −63.05 | 0.32 | −57.90 | 0.61 | −13.45 | 1.57 | −48.82 | 0.63 | −50.55 | 0.40 | −59.31 | 0.52 |
| Hydroxychloroquine | −62.66 | 0.86 | −64.86 | 1.25 | −20.44 | 3.08 | −53.26 | 0.71 | −45.68 | 1.06 | −67.91 | 1.07 |
| Chloroquine | −59.17 | 0.85 | −57.90 | 1.07 | −15.04 | 3.71 | −47.22 | 1.17 | −41.10 | 0.96 | −62.53 | 1.85 |
| Boceprevir | −58.78 | 4.05 | −59.43 | 0.80 | −11.41 | 4.15 | −41.46 | 1.41 | −34.82 | 1.07 | −61.26 | 1.80 |
| Oseltamivir | −48.68 | 0.51 | −44.43 | 0.45 | −26.16 | 3.82 | −37.56 | 0.63 | −40.48 | 1.25 | −47.34 | 0.48 |
| Ganciclovir | −48.13 | 0.81 | −44.73 | 0.55 | −23.18 | 0.80 | −41.17 | 0.62 | −36.56 | 0.48 | −45.90 | 0.25 |
| Ribavirin | −40.96 | 0.03 | −40.13 | 0.75 | −20.79 | 0.19 | −34.76 | 0.08 | −32.89 | 0.02 | −39.01 | 0.03 |
| Acyclovir | −40.94 | 0.77 | −37.72 | 0.72 | −19.23 | 1.15 | −38.45 | 0.22 | −29.47 | 0.52 | −43.91 | 0.04 |
| Favipiravir | −30.58 | 0.03 | −30.56 | 0.01 | −14.26 | 0.13 | −32.12 | 0.01 | −21.65 | 0.04 | −33.28 | 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintilie, L.; Tanase, C.; Mohapatra, R.K. Molecular Docking Studies on Synthetic Therapeutic Agents for COVID-19. Chem. Proc. 2021, 3, 46. https://doi.org/10.3390/ecsoc-24-08352
Pintilie L, Tanase C, Mohapatra RK. Molecular Docking Studies on Synthetic Therapeutic Agents for COVID-19. Chemistry Proceedings. 2021; 3(1):46. https://doi.org/10.3390/ecsoc-24-08352
Chicago/Turabian StylePintilie, Lucia, Constantin Tanase, and Ranjan Kumar Mohapatra. 2021. "Molecular Docking Studies on Synthetic Therapeutic Agents for COVID-19" Chemistry Proceedings 3, no. 1: 46. https://doi.org/10.3390/ecsoc-24-08352
APA StylePintilie, L., Tanase, C., & Mohapatra, R. K. (2021). Molecular Docking Studies on Synthetic Therapeutic Agents for COVID-19. Chemistry Proceedings, 3(1), 46. https://doi.org/10.3390/ecsoc-24-08352