2. Materials and Methods
p-Tert-butylcalix[4]arene (1) was synthesized using a reported protocol [
11]. KOH, K
2CO
3, 4-iodobenzyl bromide, copper(I) iodide, tetrabutylammonium fluoride hydrate, and nitrobenzene were purchased from Aldrich. 18-Crown-6, cyclohexene, ethynyltrimethylsilane, and palladium(II) acetate were purchased from Fluka. Triphenylphosphine was purchased from Merck and recrystallized from
n-hexane. Triethylamine was purchased from Riedel-de-Haën and used as received. Unless otherwise stated the reagents were used as received. Analytical thin-layer chromatography (TLC) was performed on Merck Kieselgel 60, F-254 silica-gel, 0.2-mm-thick plates. Fourier transform infrared (FTIR) spectra were measured on a Bruker Vertex 70.
1H NMR (400 MHz) and H-decoupled
13C NMR (100 MHz) were recorded on a Bruker ARX 400 spectrometer with CDCl
3 as the solvent and tetramethylsilane as the internal standard. The mechanosynthesis was performed on a PM100 Planetary Ball Mill (Retsch) using a 50 mL stainless-steel or zirconium oxide reactor and 200 stainless-steel or zirconium oxide balls with 5 mm of diameter.
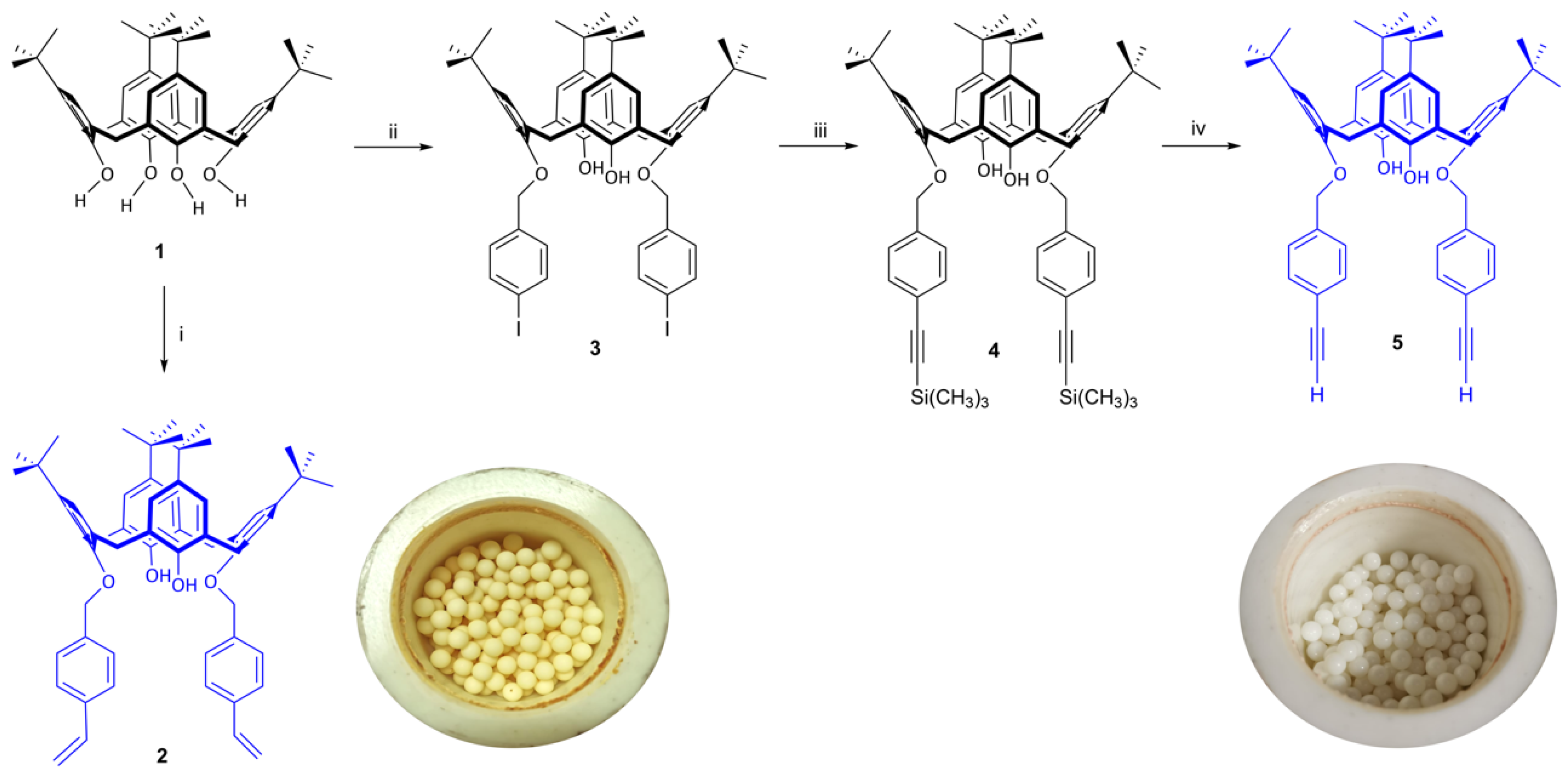
2.1. 25,27-Bis-(4-vinyl-benzyloxy)-26,28-dihydroxy-p-tert-butylcalix[4]arene (2)
Calixarene 1 (20 mg, 0.027 mmol, 1.0 equiv.), KOH (3.787 mg, 0.0675 mmol, 2.5 equiv.), 4-vinylbenzyl chloride (9.8 µL, 0.0621 mmol, 2.3 equiv.), nitrobenzene (0.4 μL, 0.027 mmol, 1.0 equiv.) and 18-crown-6 (14.5 μL, 0.0675 mmol, 2.5 equiv.) were added to a 50 mL zirconium oxide reactor containing 200 zirconium oxide balls. The reactor was then adjusted into the planetary ball mill. The mixture was ground for 60 h, at 500 rpm, with rotation inversion cycles of 30 min. (2.5 min. pause between inversion cycles). After this period the reactor was opened and the mixture was analyzed by TLC (dichloromethane/hexane 1:1), which showed a mixture of 1 and 2. The obtained yellowish resin was then dissolved in 50 mL of dichloromethane and filtered by gravity to remove 18-crown-6. After solvent removal to dryness, the yellow residue was dissolved in 50 mL of 5% HCl and extracted with chloroform (3 × 50 mL). The organic phases were collected, dried over anhydrous MgSO4, and after solvent removal to dryness the product was dried under vacuum. Calixarene 2 was further recrystallized from chloroform/n-propanol and obtained as an off-white solid in 10% yield (2.2 mg). FT-IR (KBr) ν (cm−1): 3430 (s, OH) 3150 (w, =CH2), 1629 (m, C=C), 993, 907 (m, H2C=CH); 1H NMR (CDCl3) δ (ppm): 10.37 (s, 4H, ArOH), 7.64 (4H, d, vinyl-ArH, J = 8.0 Hz) 7.40 (4H, d, vinyl-ArH, J = 8.0 Hz), 7.08 (s, 8H, ArH), 7.05 (4H, s, ArH), 6.80 (4H, s, ArH), 6.77 (1H, dd, CH=CH2, J = 10.6 and 17.6 Hz), 5.79 (1H, d, CH=CH2, J = 17.6 Hz), 5.27 (1H, d, CH=CH2, J =10.6 Hz), 5.04 (4H, s ArOCH2Ar), 4.28 (dd, 8H, ArCH2Ar, J = 13.4 Hz), 3.52 (d, 4H, ArCH2Ar, J = 13.9 Hz), 3.27 (4H, d, ArCH2Ar, J = 13.1 Hz), 1.27 (18H, s, C(CH3)3), 1.24 (s, 36H, C(CH3)3), 0.94 (18H, s, C(CH3)3).
2.2. 5,11,17,23-Tetrakis(1,1-dimethylethyl)-25,27-bis(4-iodobenzyloxy)-26,28-dihydroxycalix[4]arene (3)
Calixarene 1 (50 mg, 0.068 mmol, 1.0 equiv.), K2CO3 anhydrous (freshly flared) (35.45 mg, 2.57 mmol, 3.8 equiv.), 4-iodobenzyl bromide (50.19 mg, 0.161 mmol, 2.38 equiv.) were added to a 50 mL zirconium oxide reactor containing 200 zirconium oxide balls. The reactor was then adjusted into the planetary ball mill. The mixture was ground for 7 h, at 500 rpm, with rotation inversion cycles of 15 min. (5 s pause between inversion cycles). After this period the reactor was opened and the mixture was analyzed by TLC (dichloromethane/petroleum ether 1:1), which showed total consumption of 1 and the formation of 3. The obtained brown residue was then dissolved in 50 mL of dichloromethane and filtered by gravity. After solvent removal to dryness, the light brown residue was extracted with water (3 × 50 mL). The organic phases were collected, dried over anhydrous MgSO4, and after solvent removal to dryness the product was dried under vacuum to give a beige solid. Calixarene 3 was further recrystallized from chloroform/methanol and obtained as a white solid in 27.2% yield (19.8 mg). FT-IR (KBr) ν (cm−1): 3530, 3409 (s, OH), 2961 (s, CH, (CH3)3), 1594 (m, C=C), 1482 (s, CH, CH2), 871, 800 (s, =CH). 1H NMR (CDCl3) δ (ppm): 7.72 (4H, d, I-ArH, J = 7.4 Hz), 7.41 (4H, s, I-ArH), 7.10 (2H, s, ArOH), 7.04 (4H, s, ArH), 6.77 (4H, s, I-ArH), 4.99 (4H, s, ArOCH2Ar), 4.22 (4H, d, ArCH2Ar, J = 13.0 Hz), 3.27 (4H, d, ArCH2Ar, J = 13.0 Hz), 1.29 (18H, s, C(CH3)3), 0.93 (18H, s, C(CH3)3).
2.3. 5,11,17,23-Tetrakis(1,1-dimethylethyl)-25,27-bis[4-(trimethylsilylethynyl)benzyloxy]-26,28-dihydroxycalix[4]arene (4)
Calixarene 3 (200 mg, 0.185 mmol, 1.0 equiv.), Et3N (740 μL, 4 mL/mmol), Pd(AcO)2 (2.077 mg, 9.25 μmol, 5 mol%), CuI (3.5 mg, 18.5 μmol, 10 mol%), PPh3 (recrystallized from n-hexane) (4.85 mg, 18.5 μmol, 10 mol%), ethinyltrimethylsilane (62 μL, 0.44 mmol, 2.4 equiv.), MgSO4 anhydrous (1 g, 5 equiv./mg, reaction milling auxiliary), and cyclohexene (40 μL, 0.2 μL/mg, catalyst milling auxiliary) were added to a 50 mL zirconium oxide reactor containing 200 zirconium oxide balls. The reactor was then adjusted into the planetary ball mill. The mixture was ground for 8 h, at 500 rpm, with rotation inversion cycles of 30 min. (2.5 min. pause between inversion cycles). After this period the reactor was opened and the mixture was analyzed by TLC (dichloromethane/hexane 1:4, double elution), which showed a mixture of 3 and 4. The obtained light brown residue was then dissolved in 50 mL of dichloromethane and filtered by gravity to remove MgSO4. After solvent removal to dryness, the mixture was redissolved in 50 mL of dichloromethane and extracted with a saturated NH4Cl solution (2 × 50 mL) and water. The organic phases were collected, dried over anhydrous MgSO4, and after solvent removal to dryness the product was dried under vacuum. Calixarene 4 was further recrystallized from dichloromethane/methanol and obtained as a light brown solid in 36.9% yield (69.7 mg). FT-IR (KBr) ν (cm−1): 3528, 3427, 3412 (s, OH), 2960 (s, CH, (CH3)3), 2158 (C≡C), 1605, 1592 (m, C=C), 1485 (s, CH, CH2), 1250 (s, Si(CH3)3), 872, 867, 806 (s, =CH). 1H NMR (CDCl3) δ (ppm): 7.72 (d, 4H, I-ArH, J = 8.3 Hz), 7.49 (s, 8H, (CH3)3SiC≡C-ArH), 7.41 (d, 4H, I-ArH, J = 8.1 Hz), 7.10 (d, 4H, ArOH), 7.04 (d, 8H, ArH), 6.77 (s, 4H, I-ArH), 6.75 (s, 4H, ArH), 5.01 (s, 4H, ArOCH2Ar), 4.99 (s, 4H, ArOCH2Ar), 4.20 (dd, 8H, ArCH2Ar, J = 13.1 Hz), 3.23 (dd, 8H, ArCH2Ar, J = 13.1 Hz), 1.28 (d, 36H, C(CH3)3), 0.94 (s, 36H, C(CH3)3), 0.27 (s, 18H, Si(CH3)3).
2.4. 5,11,17,23-Tetrakis(1,1-dimethylethyl)-25,27-bis(4-ethynylbenzyloxy)-26,28-dihydroxycalix[4]arene (5)
Calixarene 4 (20 mg, 0.19 mmol, 1.0 equiv.) and n-Bu4NF (11.492 mg, 0.043 mmol, 2.1 equiv.) were added to a 50 mL zirconium oxide reactor containing 200 zirconium oxide balls. The reactor was then adjusted into the planetary ball mill. The mixture was ground for 1 h, at 500 rpm, with rotation inversion cycles of 15 min. (5 s pause between inversion cycles). After this period the reactor was opened and the mixture was analyzed by TLC (dichloromethane/hexane 3:1), which showed a total consumption of 4 and the formation of 5. The obtained white residue was then dissolved in 25 mL of dichloromethane and washed with a solution of 10% HCl (25 mL) and water. The organic phases were collected, dried over anhydrous MgSO4, and after solvent removal to dryness the product was dried under vacuum. Calixarene 5 was further recrystallized from dichloromethane/methanol and obtained as a foamy brown-orange solid in 58.2% yield (10 mg). FT-IR (KBr) ν (cm−1): 3404 (s, OH), 3304, 3282, 3257 (s, ≡CH), 2961 (s, CH, C(CH3)3, 2107 (w, C≡C), 1598 (m, C=C), 1485 (s, CH, CH2), 873, 820, 735 (s, =CH), 618 (s, C≡C). 1H NMR (CDCl3) δ (ppm): 7.60 (d, 4H, C≡C-ArH, J = 8.0 Hz), 7.52 (d, 4H, C≡C-ArH, J = 8.0 Hz), 7.12 (s, 2H, ArOH), 7.04 (s, 4H, ArH), 6.77 (s, 4H, ArH), 5.04 (s, 4H, ArOCH2Ar), 4.24 (d, 4H, ArCH2Ar, J = 13.0 Hz), 3.27 (d, 4H, ArCH2Ar, J = 13.0 Hz), 3.11 (s, 2H, ≡CH), 1.29 (s, 18H, C(CH3)3), 0.93 (s, 18H, C(CH3)3).
3. Results and Discussion
The green synthesis of functional calix[4]arenes was performed using a mechanochemical approach. Overall, the solventless synthetic route showed important advantages over conventional protocols. In this work, starting from
p-
tert-butylcalix[4]arene
1, two functional calixarenes monomers, bearing aryl vinyl or aryl ethynyl pending units, were successfully prepared (
Scheme 1).
The reactions were conducted in a planetary ball mill using both stainless-steel and zirconium oxide reactors, and the reactor was found to influence the reaction outcome. In most cases, a low yield or no reaction was observed when the stainless-steel was used.
Calixarene
2 [
12] was synthesized in one step using 4-vinylbenzyl chloride, KOH as a base, and nitrobenzene as a milling auxiliary (equimolar amount). It is well known that K
+ ions can be easily trapped in the calixarene cavity, altering the cone configuration, and ultimately decreasing the lower rim reactivity. Therefore, the effect of the addition of 18-crown-6, a ligand with an affinity for potassium cations, was investigated. Although a lower yield was observed in comparison with the optimized conventional protocol, 10% vs. 48% yield, the reaction time was reduced from 168 h (reflux in acetonitrile) to 60 h (milling at 500 rpm) (
Table 1).
Further reaction optimization to increase the reaction yield is undergoing. Surprisingly, the base selection was also found to be critical. When K2CO3 (conventional route base) was used, only traces of calixarene 2 were obtained.
Similarly, calixarene
5 [
13] was prepared in three steps (
Table 1). First, calixarene
1 was reacted with 4-iodobenzyl bromide using K
2CO
3 anhydrous as a base to give calixarene
3 after 7 h of milling at 500 rpm. The yield is much lower than the conventional reaction, 27.2%
vs. 81.1% yield; however, the reaction time was reduced from 24 to 7 h under solventless conditions. Next calixarene
4 was obtained from calixarene
3 via a Sonogashira–Hagihara coupling in 8 h of milling at 500 rpm. The reaction yield was optimized by the introduction of MgSO
4 anhydrous as a grinding auxiliary and cyclohexene as an additive. Olefins, and 1,5-cyclooctadiene (COD) in particular, have been reported to remarkably accelerate palladium-catalyzed cross-coupling solid-state reactions by acting as efficient molecular dispersants [
14]. In this protocol, we chose cyclohexene, a much cheaper and less toxic olefin. Under these conditions, calixarene
4 was obtained in 36.9% yield after 8 h of grinding, which is quite good if compared with conventional conditions (68.1%), using tetrahydrofuran under reflux for 24 h. Finally, calixarene
5 was obtained in 15 min. by removal of the TMS protecting group using
n-Bu
4NF. The mechanochemical-assisted deprotection, in this case, was even higher than that obtained under conventional conditions (58.2% vs. 49.1% yield), avoiding the use of tetrahydrofuran.



 ,
, 




{kind=link}