
A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy †


Abstract
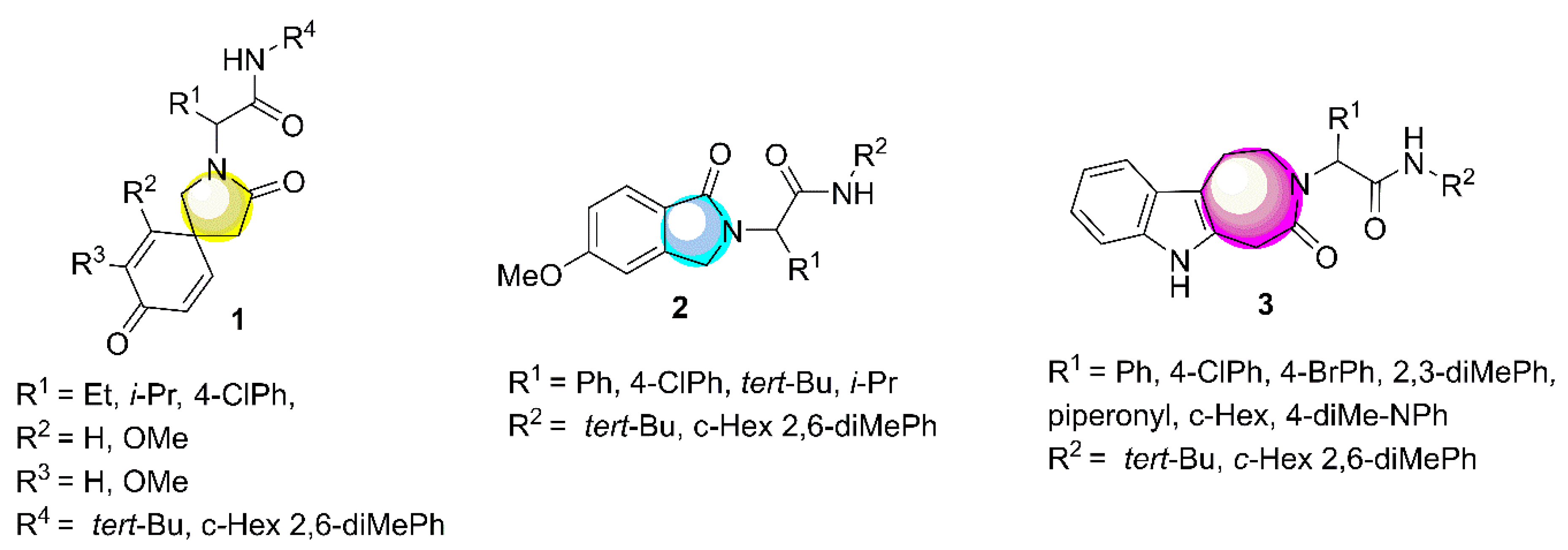
:1. Introduction
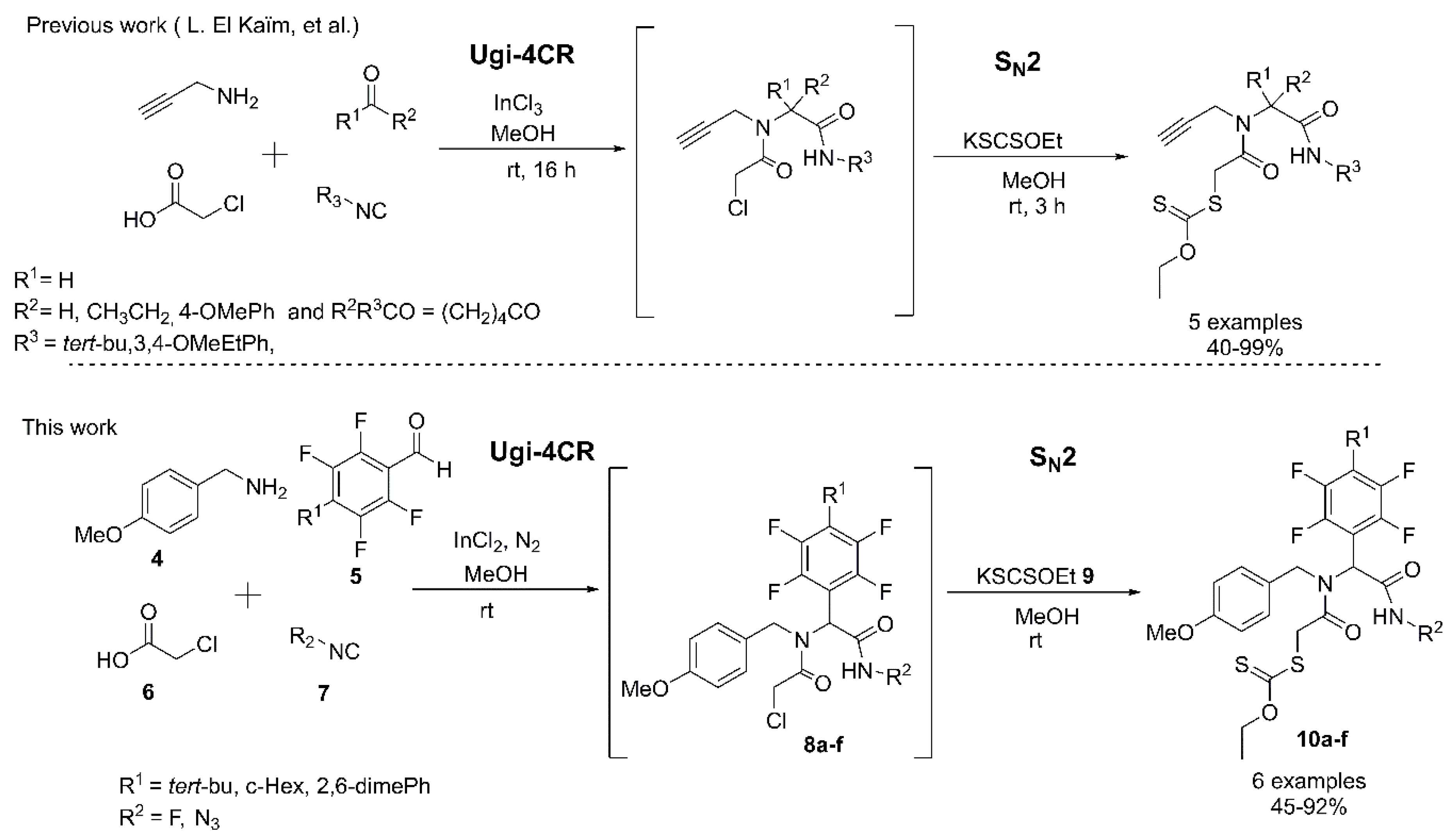
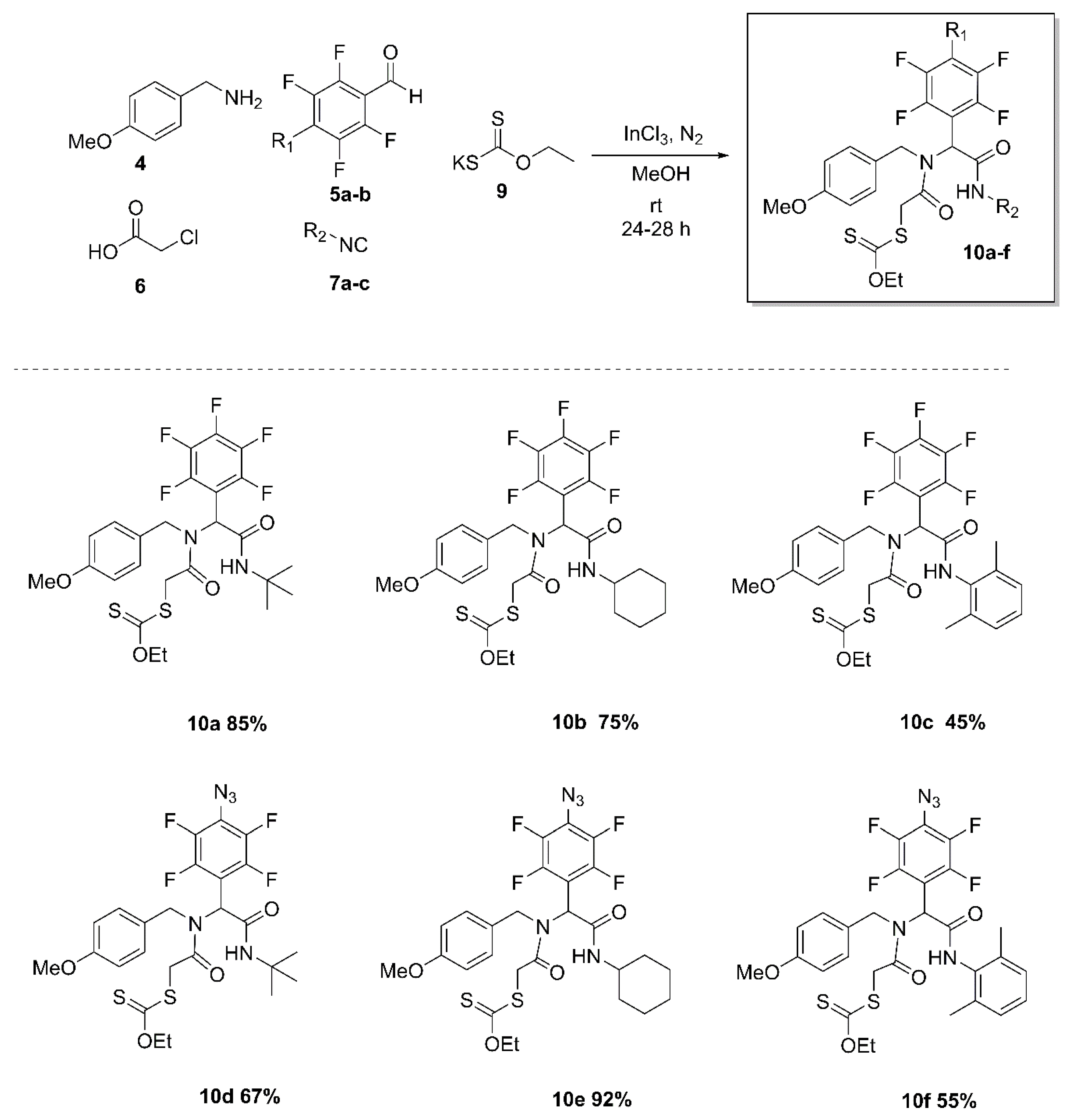
2. Results and Discussion
3. Experimental Section
3.1. General Information, Instrumentation, and Chemicals
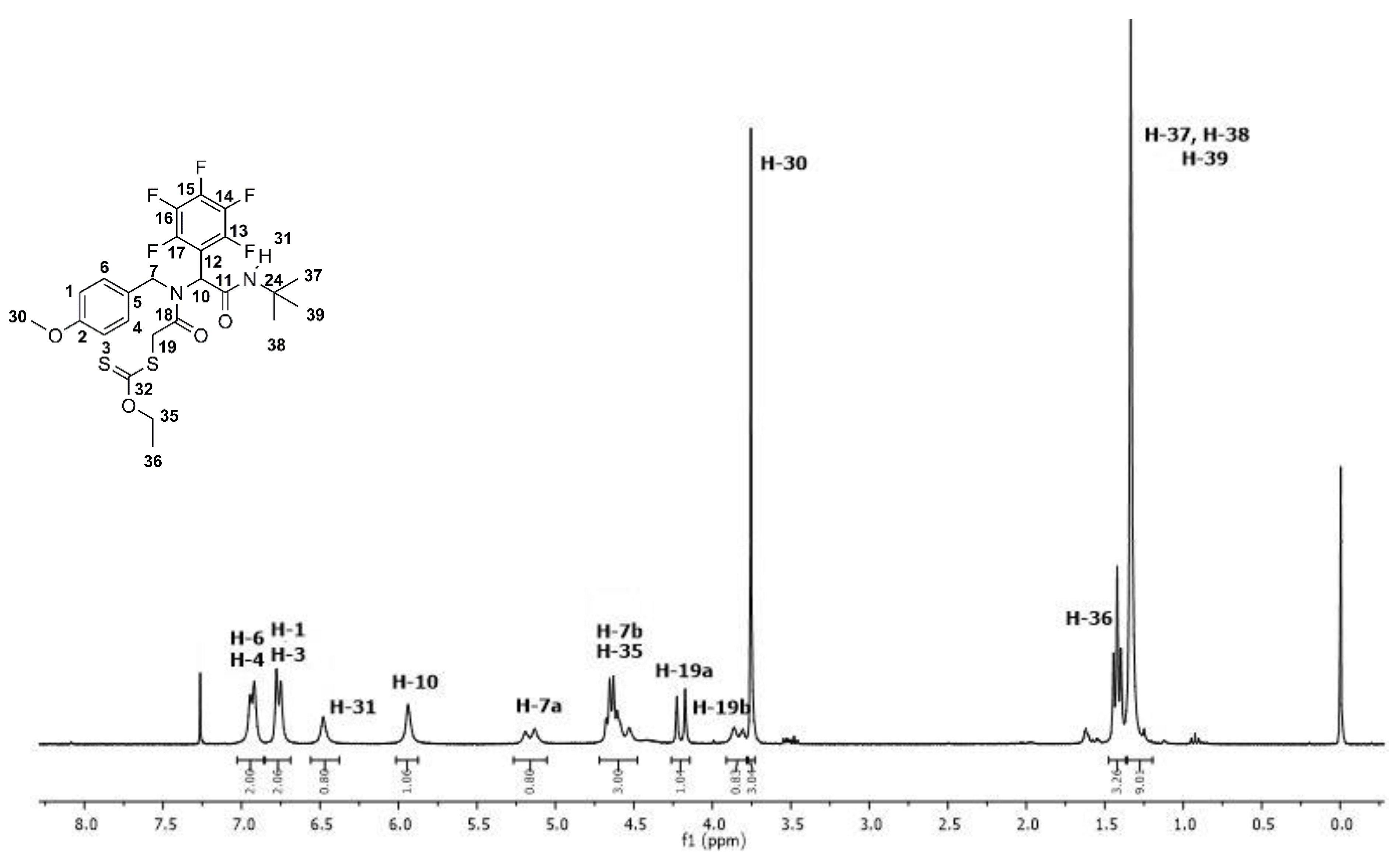
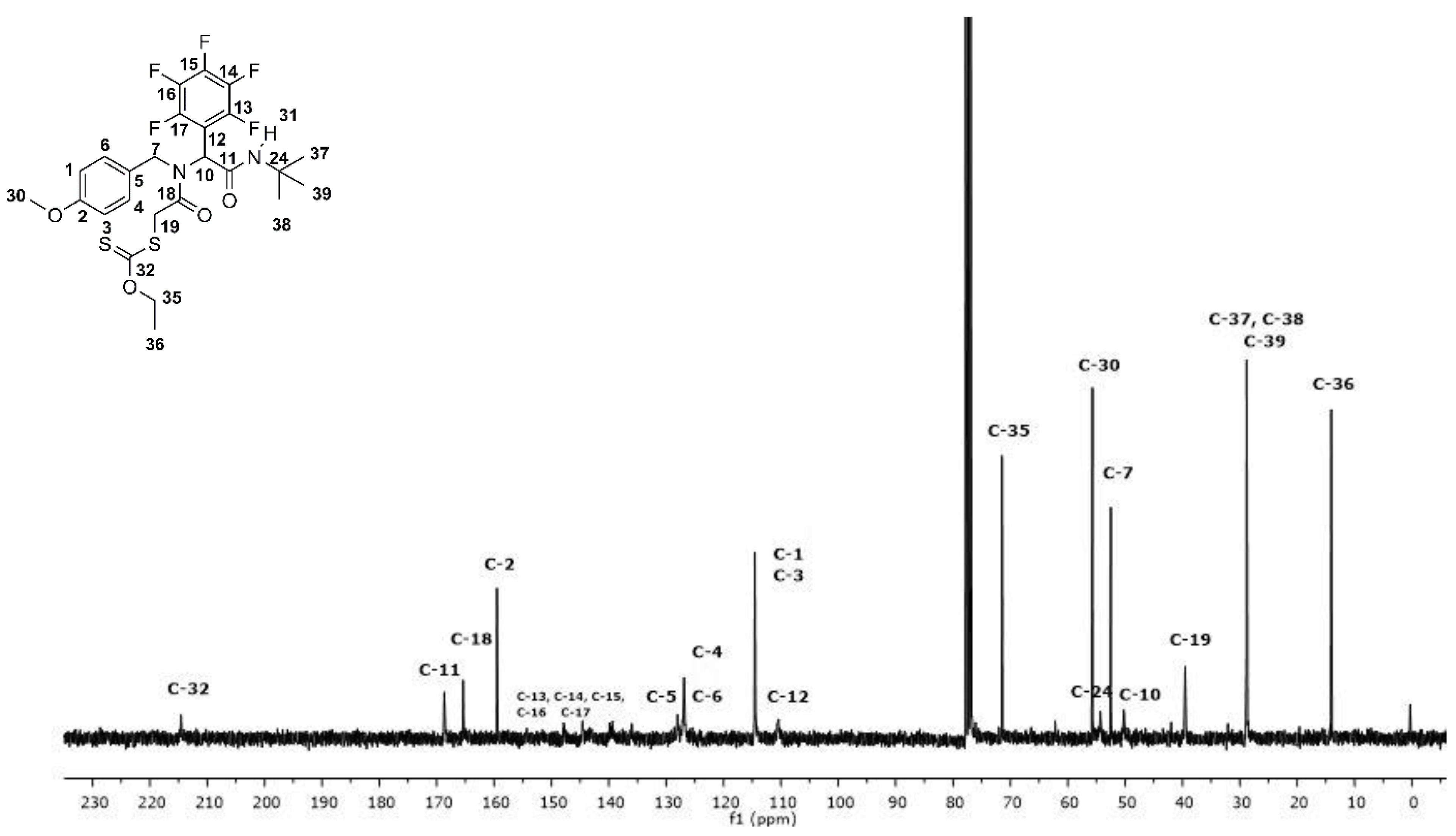
3.2. Synthesis and Characterization of the Fluoro α-Acylamino Amide-Xanthates 10a–f
3.2.1. S-(2-((2-(tert-Butylamino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10a)
3.2.2. S-(2-((2-(Cyclohexylamino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10b)
3.2.3. S-(2-((2-((2,6-Dimethylphenyl)amino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10c)
3.2.4. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-(tert-butylamino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10d)
3.2.5. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-(cyclohexylamino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10e)
3.2.6. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-((2,6-dimethylphenyl)amino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate e (10f)
4. Conclusions
Author Contributions
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Quiclet-Sire, B.; Zard, S.Z. Powerful Carbon-Carbon Bond Forming Reactions Based on a Novel Radical Exchange Process. Chem. Eur. J. 2006, 12, 6002–6016. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Montaño, R.; Ibarra-Rivera, T.; Kaïm, L.; Miranda, L. Efficient Synthesis of Azaspirodienones by Microwave-Assisted Radical Spirocyclization of Xanthate-Containing Ugi Adducts. Synthesis 2010, 8, 1285–1290. [Google Scholar] [CrossRef]
- Rentería-Gómez, A.; Islas-Jácome, A.; Jiménez-Halla, J.O.C.; Gámez-Montaño, R. Regiospecific synthesis of 1-acetamide-5-methoxy-2-oxindoles in two steps: (Ugi-SN2)/xanthate mediated free radical cyclization. Tetrahedron Lett. 2014, 55, 6567–6570. [Google Scholar] [CrossRef]
- Gordillo-Cruz, R.E.; Rentería-Gómez, A.; Islas-Jácome, A.; Cortes-García, C.J.; Díaz-Cervantes, E.; Robles, J.; Gámez-Montaño, R. Synthesis of 3-tetrazolylmethyl-azepino [4,5-b]indol-4-ones in two reaction steps: (Ugi-azide/N-acylation/SN2)/free radical cyclization and docking studies to a 5-Ht6 model. Org. Biomol. Chem. 2013, 11, 6470. [Google Scholar] [CrossRef]
- Chaturvedi, D.; Ray, S. An Efficient, One-Pot, Triton-B Catalyzed Synthesis of O-Alkyl-S-methyl Dithiocarbonates. Chem. Mon. 2006, 137, 1219–1223. [Google Scholar] [CrossRef]
- El Kaïm, L.; Grimaud, L.; Miranda, L.D.; Vieu, E.; Cano-Herrera, M.-A.; Perez-Labrada, K. New xanthate-based radical cyclization onto alkynes. Chem. Commun. 2010, 46, 2489–2491. [Google Scholar] [CrossRef] [PubMed]
- Kaïm, L.E.; Grimaud, L.; Miranda, L.D.; Vieu, E. Ugi/xanthate cyclizations as a radical route to lactam scaffolds. Tetrahedron Lett. 2006, 47, 8259–8261. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent c | Additive | T (°C) | Time | Yield (%) f |
| 1 a | MeOH | --- | rt | 7 days | 72 |
| 2 a | MeOH | InCl3 d | rt | 3 h | 35 |
| 3 a | MeOH | InCl3 d | rt | 24 h | 66 |
| 4 a | MeOH | InCl3 d | 50 °C MW | 1 h | nd |
| 5 b | MeOH | InCl3 e | rt | 24 h | 75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rentería-Gómez, M.A.; Ibarra-Rivera, T.R.; Gámez-Montaño, R. A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy. Chem. Proc. 2021, 3, 59. https://doi.org/10.3390/ecsoc-24-08421
Rentería-Gómez MA, Ibarra-Rivera TR, Gámez-Montaño R. A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy. Chemistry Proceedings. 2021; 3(1):59. https://doi.org/10.3390/ecsoc-24-08421
Chicago/Turabian StyleRentería-Gómez, Manuel A., Tannya R. Ibarra-Rivera, and Rocío Gámez-Montaño. 2021. "A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy" Chemistry Proceedings 3, no. 1: 59. https://doi.org/10.3390/ecsoc-24-08421
APA StyleRentería-Gómez, M. A., Ibarra-Rivera, T. R., & Gámez-Montaño, R. (2021). A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy. Chemistry Proceedings, 3(1), 59. https://doi.org/10.3390/ecsoc-24-08421