
Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search †



Abstract
:1. Introduction
2. Methods
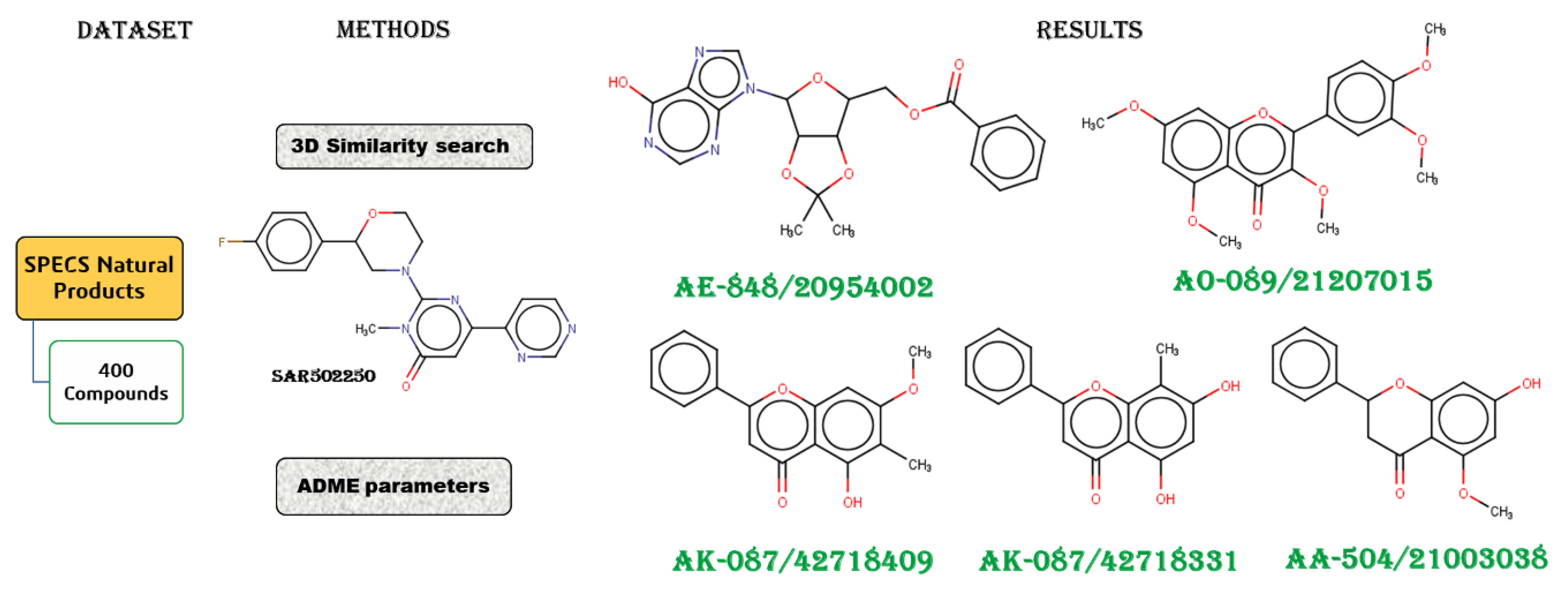
2.1. Workflow
2.2. Dataset Preparation
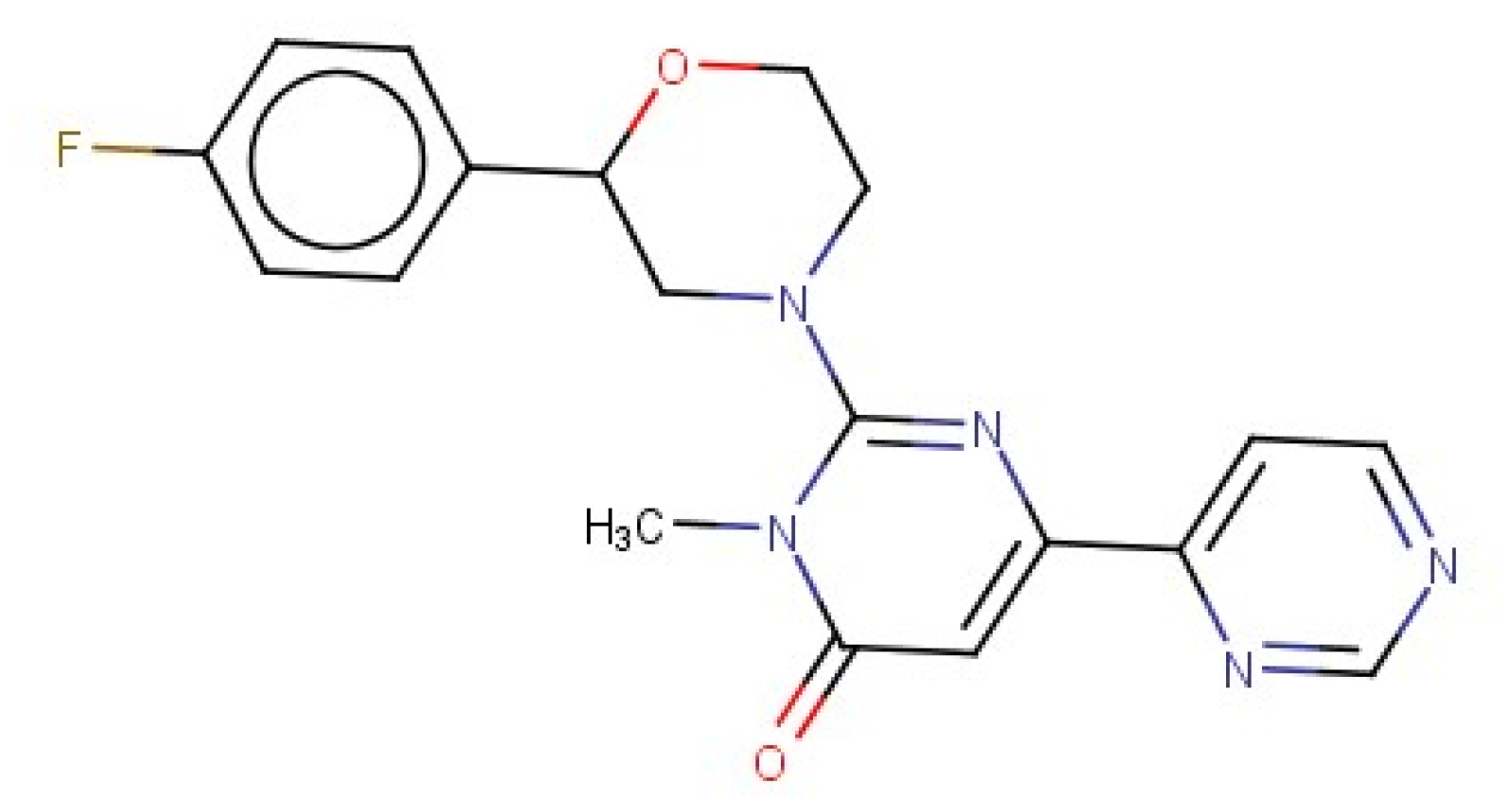
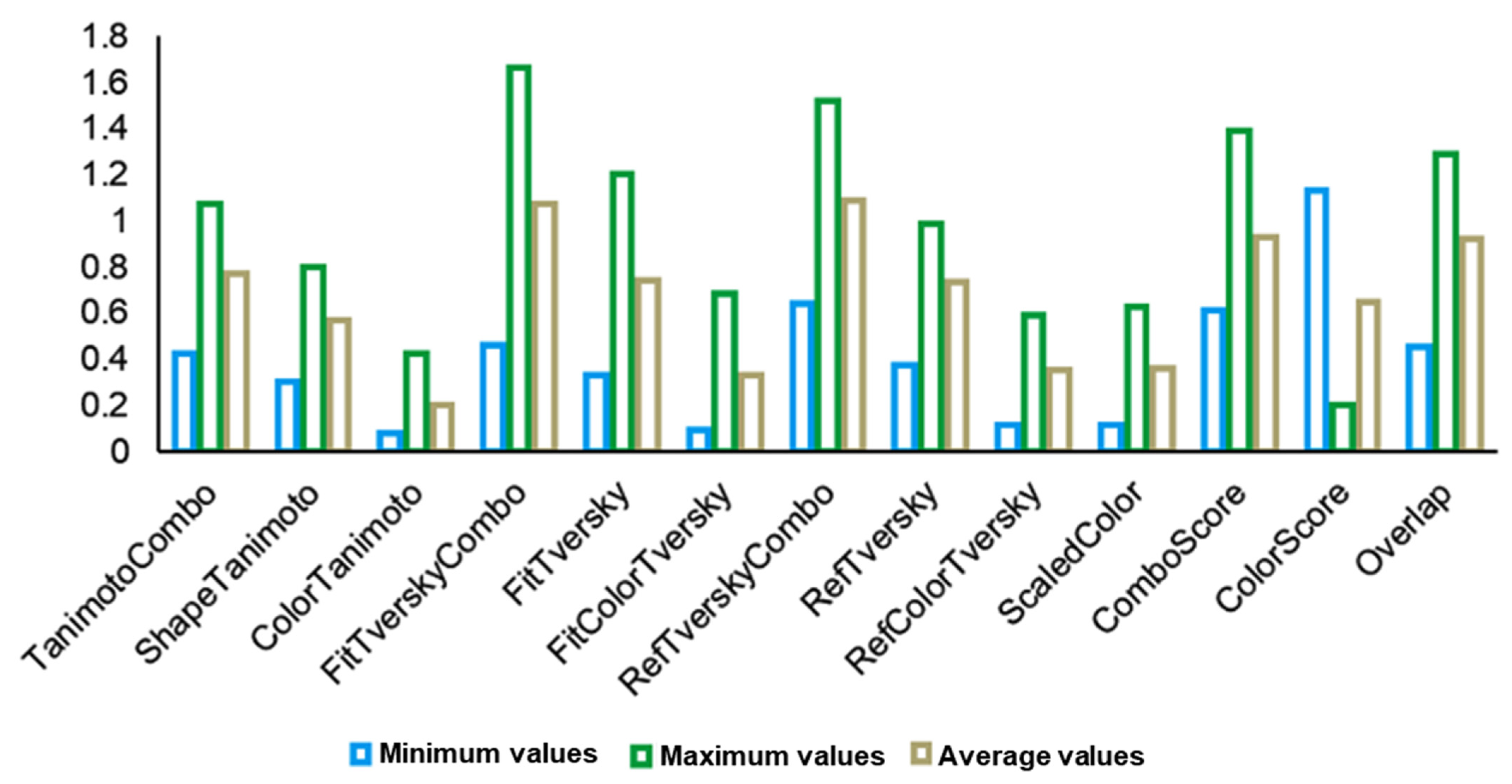
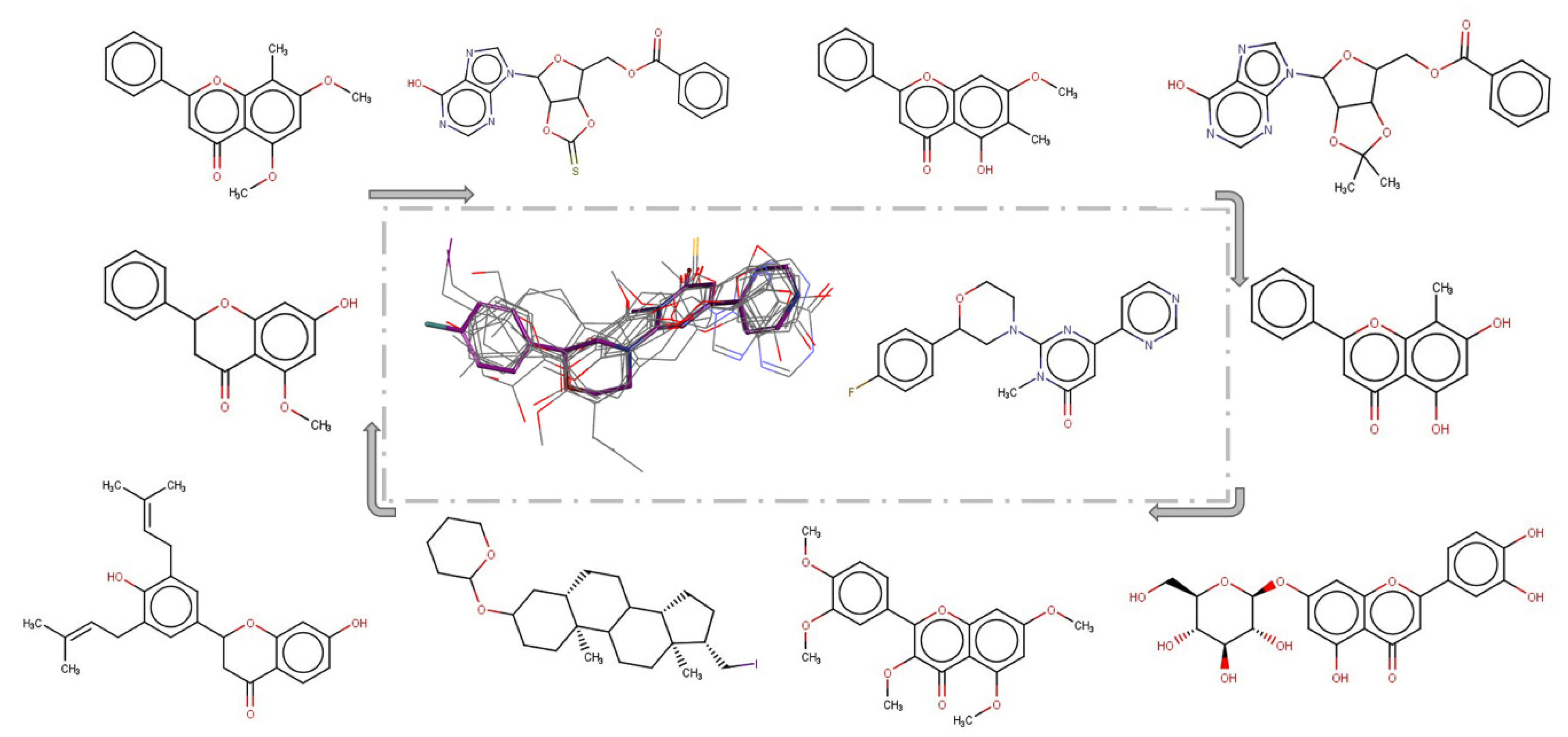
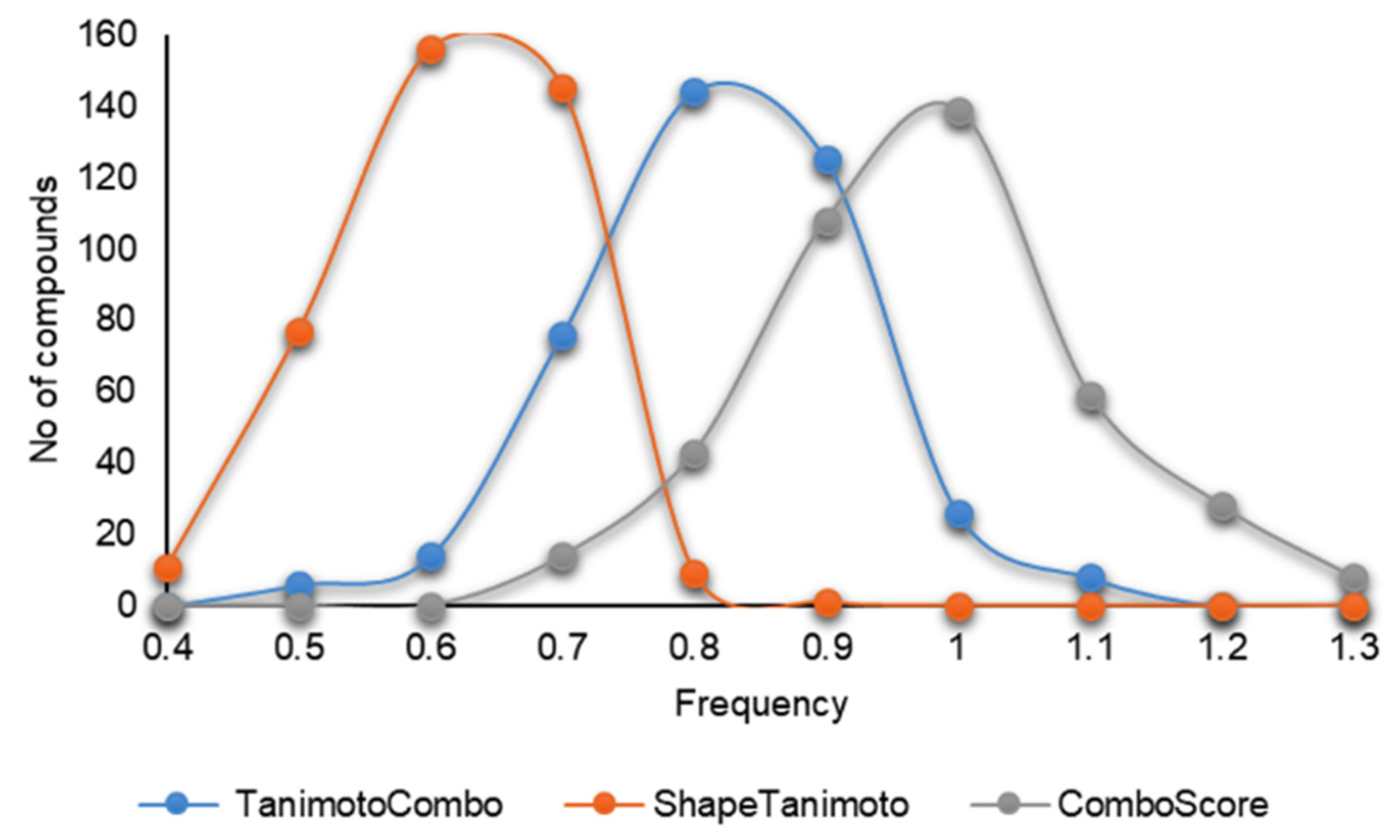
2.3. 3D Similarity Search
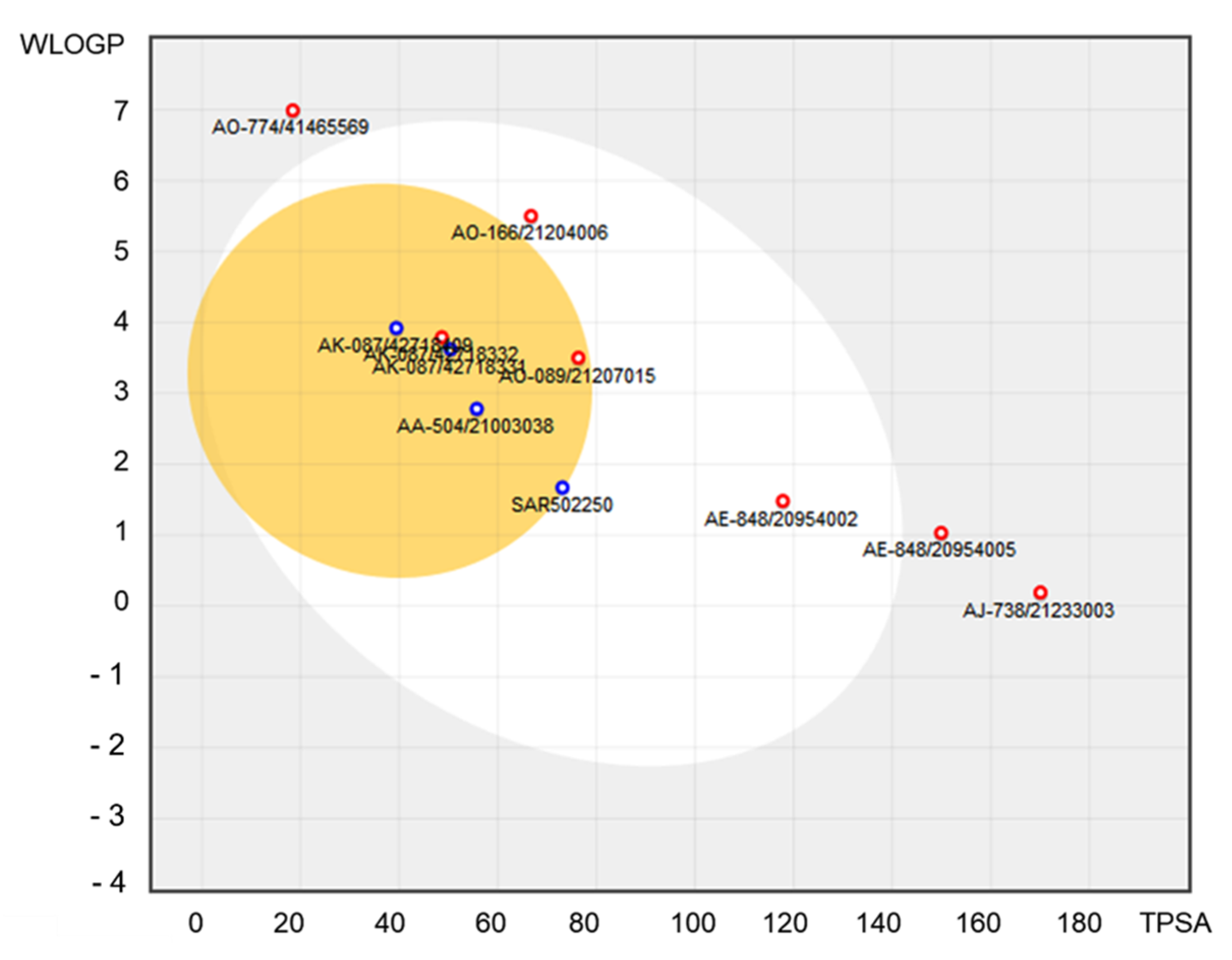
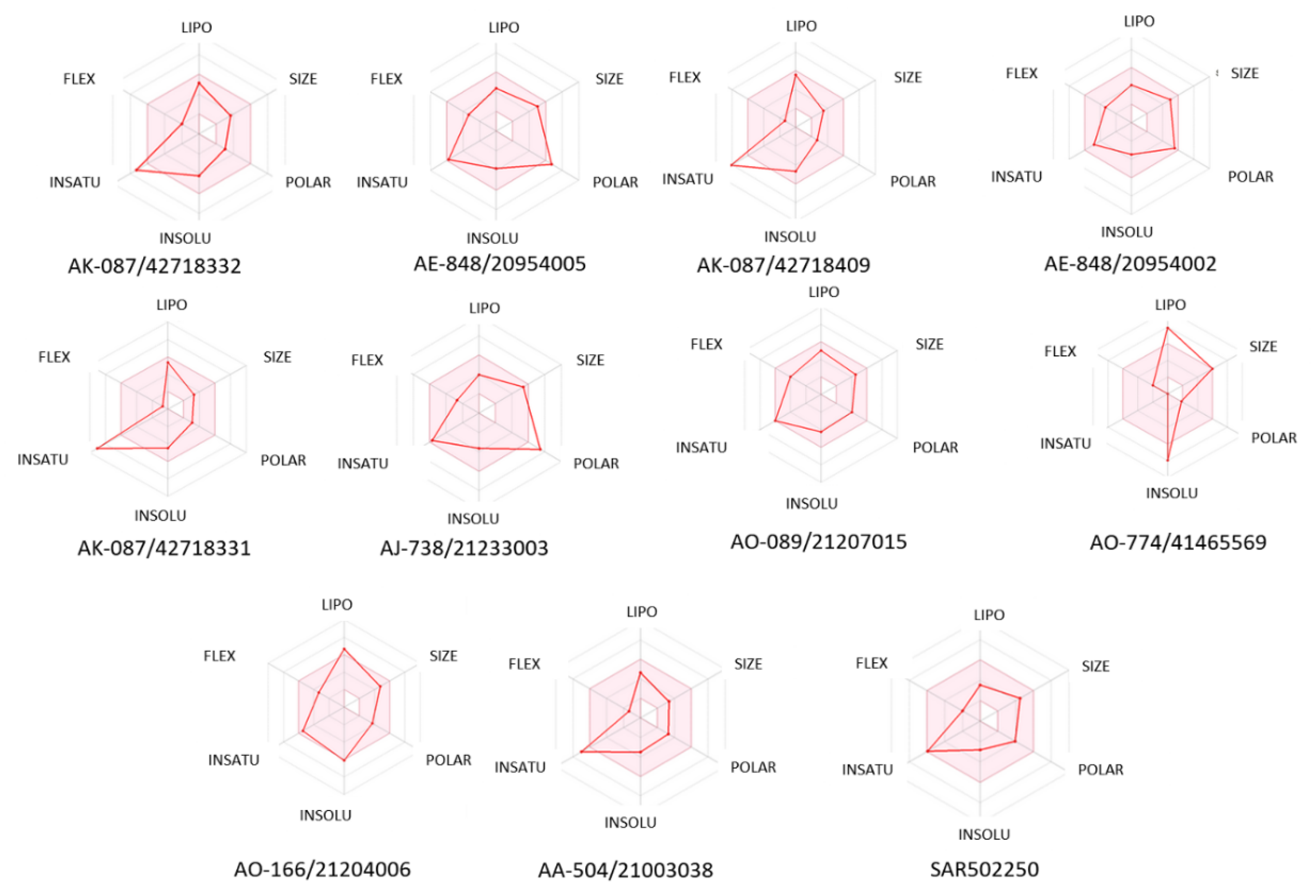
2.4. ADME and Toxicity-Related Risk Profiles
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.J.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Nabben, M.; Neumann, D. GSK-3 Inhibitors: Anti-Diabetic Treatment Associatedwith Cardiac Risk? Cardiovasc. Drugs 2016, 30, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef]
- Ivan, D.; Crisan, L.; Funar-Timofei, S.; Mracec, M. A quantitative structure-activity relationships study for the anti-HIV-1 activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives using the multiple linear regression and partial least squares methodologies. J. Serb. Chem. Soc. 2013, 78, 495–506. [Google Scholar] [CrossRef]
- Smith, D.G.; Buffet, M.; Fenwick, A.E.; Haigh, D.; Ife, R.J.; Saunders, M.; Slingsby, B.P.; Stacey, R.; Ward, R.W. 3-Anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2001, 11, 635–639. [Google Scholar] [CrossRef]
- Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.L.; Myrianthopoulos, V.; Mikros, E.; Tarricone, A.; Musacchio, A.; Roe, S.M.; Pearl, L.; Leost, M.; et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004, 47, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Lauenroth, K.; Leost, M.; Meijer, L.; Lemcke, T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2004, 14, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Mouawad, L.; Legraverend, M. Novel 8-arylated purines as inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2010, 45, 3389–3393. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Gaisina, I.N.; El-Khodor, B.F.; Ramboz, S.; Makhortova, N.R.; Rubin, L.L.; Kozikowski, A.P. Identification of a maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitor, BIP-135, that prolongs the median survival time of D7 SMA KO mouse model of spinal muscular atrophy. ACS Chem. Neurosci. 2012, 3, 5–11. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef]
- Specs. Compound Management Services and Research Compounds for the Life Science Industry. Available online: https://www.specs.net/index.php (accessed on 12 February 2020).
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.S.; Grant, J.A.; Mosyak, L.; Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef]
- Schrödinger, LLC. LigPrep; Schrödinger, LLC: New York, NY, USA, 2014. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Osiris. Osiris Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 3 August 2020).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Debe, D.A.; Metz, J.T.; Brown, S.P.; Martin, Y.C.; Hajduk, P.J. Application of Belief Theory to Similarity Data Fusion for Use in Analog Searching and Lead Hopping. J. Chem. Inf. Model. 2008, 48, 941–948. [Google Scholar] [CrossRef]
- Bortolato, A.; Perruccio, F.; Moro, S. Successful Applications of In Silico Approaches for Lead/Drug Discovery; Miteva, M.A., Ed.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | AK-087/ 42718332 | AE-848/ 20954005 | AK-087/ 42718409 | AE-848/ 20954002 | AK-087/ 42718331 | AJ-738/ 21233003 | AO-089/ 21207015 | AO-774/ 41465569 | AO-166/ 21204006 | AA-504/ 21003038 | SAR502250 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BBB permeant | Yes | No | Yes | No | Yes | No | Yes | No | No | Yes | Yes |
| MW | 296.32 | 414.39 | 281.28 | 412.4 | 267.26 | 447.37 | 372.37 | 500.5 | 392.49 | 270.28 | 367.38 |
| RBN | 3 | 5 | 2 | 5 | 1 | 4 | 6 | 3 | 5 | 2 | 3 |
| MR | 85.87 | 100.26 | 79.51 | 102.12 | 75.04 | 106.24 | 100.38 | 126.33 | 116.99 | 74.02 | 100.96 |
| TPSA | 48.67 | 149.91 | 39.44 | 117.82 | 50.44 | 170.05 | 76.36 | 18.46 | 66.76 | 55.76 | 73.14 |
| XLOGP3 | 3.49 | 2.14 | 4.21 | 1.66 | 3.88 | 1.46 | 3.25 | 8.3 | 6.15 | 2.65 | 0.68 |
| WLOGP | 3.79 | 1.03 | 3.92 | 1.48 | 3.62 | 0.19 | 3.5 | 6.99 | 5.5 | 2.78 | 1.67 |
| GI absorption | High | Low | High | High | High | Low | High | Low | High | High | High |
| Lipinski #violations | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 |
| TanimotoCombo | 1.075 | 1.049 | 1.04 | 1.025 | 1.013 | 1.009 | 1.007 | 1.006 | 0.997 | 0.996 | 2 |
| ShapeTanimoto | 0.651 | 0.769 | 0.65 | 0.739 | 0.63 | 0.802 | 0.681 | 0.696 | 0.695 | 0.611 | 1 |
| ComboScore | 1.18 | 1.304 | 1.18 | 1.332 | 1.153 | 1.392 | 1.2 | 1.143 | 1.211 | 1.136 | 2 |
| Toxicity risk * | • M | • M | • M | • M | • M | • M | • M | • M | • M | • M | • M |
| • T | • T | • T | • T | • T | • T | • T | • T | • T | • T | • T | |
| • I | • I | • I | • I | • I | • I | • I | • I | • I | • I | • I | |
| • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE | • RE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisan, L.; Bora, A.; Pacureanu, L. Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search. Chem. Proc. 2021, 3, 75. https://doi.org/10.3390/ecsoc-24-08341
Crisan L, Bora A, Pacureanu L. Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search. Chemistry Proceedings. 2021; 3(1):75. https://doi.org/10.3390/ecsoc-24-08341
Chicago/Turabian StyleCrisan, Luminita, Alina Bora, and Liliana Pacureanu. 2021. "Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search" Chemistry Proceedings 3, no. 1: 75. https://doi.org/10.3390/ecsoc-24-08341
APA StyleCrisan, L., Bora, A., & Pacureanu, L. (2021). Identification of Natural Products for the Treatment of Alzheimer’s Disease: 3D Similarity Search. Chemistry Proceedings, 3(1), 75. https://doi.org/10.3390/ecsoc-24-08341