1. Introduction
Hydrazones are well known compounds that have shown potent antioxidant activities with respect to free radical scavenging. Normally, hydrazones are characterized by an imine group (azomethine), which is essential for their antioxidant activities [
1].
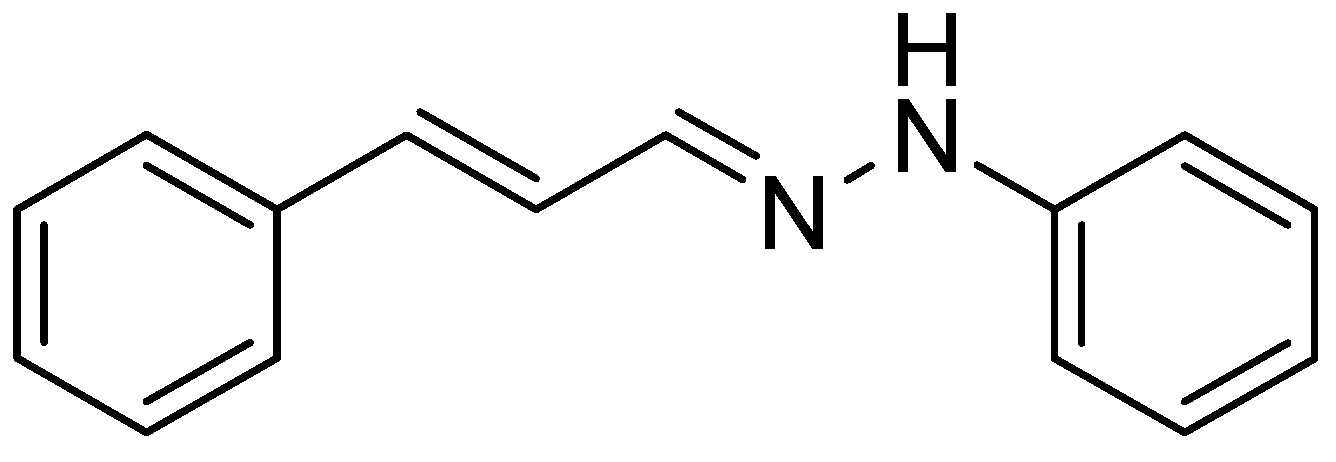
Cinnamaldehyde phenylhydrazone (
Figure 1) was synthesized from cinnamaldehyde and phenylhydrazine, and its antioxidant properties have been demonstrated [
2].
Molecular docking techniques are used to predict how a protein interacts with small molecules, such as antioxidants. This ability acts on a significant part of the protein’s dynamics that may enhance/inhibit its interaction function in terms of which molecules are targeted [
3]. The molecular docking studies can be used to model the interaction between a small molecule and a protein at the atomic level, allowing one to characterize the behavior of small molecules at the binding site of target proteins [
4]. Thus, the accurate prediction of the binding modes between the ligand and the protein is of fundamental importance in the design of small transport molecules based on modern structures. Considering that the enzymes cytochrome P450 (CP450) and NADPH oxidase (NO) are associated with the oxidative stress process, in this work, we chose these enzymes to evaluate their interaction with the antioxidant CPH. The ability of CPH to interact with these enzymes will be a measure of the capacity of inhibiting the oxidative process.
The aim of the current work was to evaluate the interaction of CP450 and NADPH oxidase NO enzymes, the main fraction of the casein, with cinnamaldehyde phenylhydrazone (CPH), using molecular docking calculations. We used a SwissDock server, applying specific scoring functions based on energy terms to obtain the best protein–ligand binding patterns and binding affinity between CPA and CP450-NADPH enzymes.
2. Computational Methodology
2.1. Cinnamaldehyde Phenylhydrazone Structure Preparation
The structure of the antioxidant ligand in this study was optimized employing the Gaussian09 program suit (Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, USA) with the hybrid density functional B3LYP and 6-31+G(d,p) basis sets.
2.2. Proteins Structure Preparation
The amino acid sequences of CP-450 and NO were obtained from the Protein Data Bank of RCSB [
5]. The structure of CP-450 corresponds to the PDB: 1OG5 (structure of human cytochrome P450 CYP2C9) with 475 residues of amino acid; and the structure of NO corresponded to the PDB: 2CDU (structure of water-forming NAD(P)H oxidase from
Lactobacillus sanfranciscensis) with 452 residues. The protonation state of the ionizable residues at pH = 7 was evaluated with the PROPKA program [
6]. The final structures were minimized with the USCF Chimera program according to the MM calculation method and were validated by the Mol-Probity server [
7].
2.3. Molecular Docking Studies
After the preparation of proteins and ligand structures, molecular docking calculations were performed by SwissDock servers [
8]. These docking studies corresponded to a system with a flexible ligand and a rigid protein. Using specific scoring functions based on energy terms, the best protein–ligand binding models were obtained. Interaction types and distances were evaluated with the USCF Chimera program and Discovery Studio Visualizer [
9].
3. Results and Discussion
3.1. Molecular Docking Calculation Conducted with CPH/CP-450
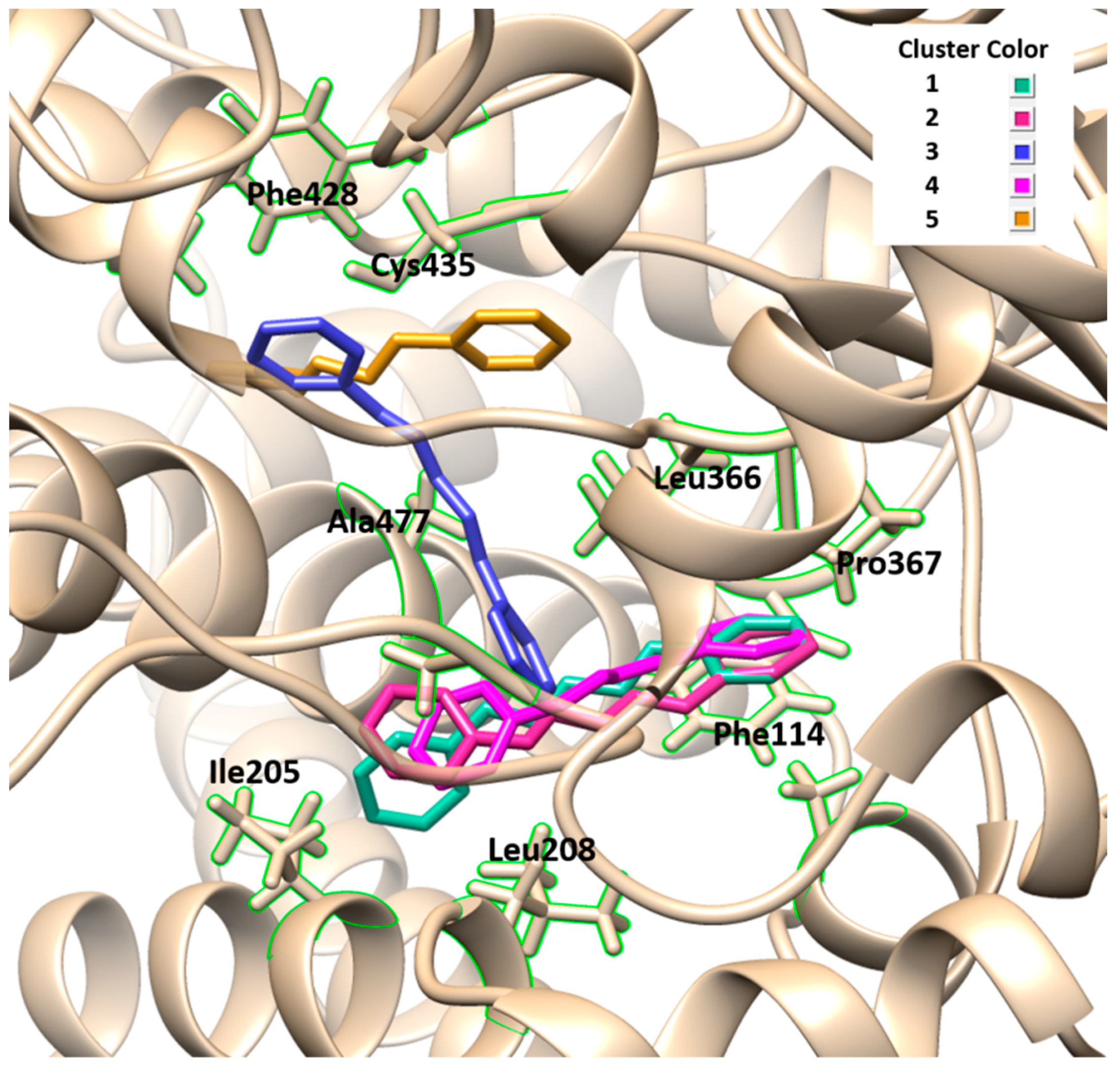
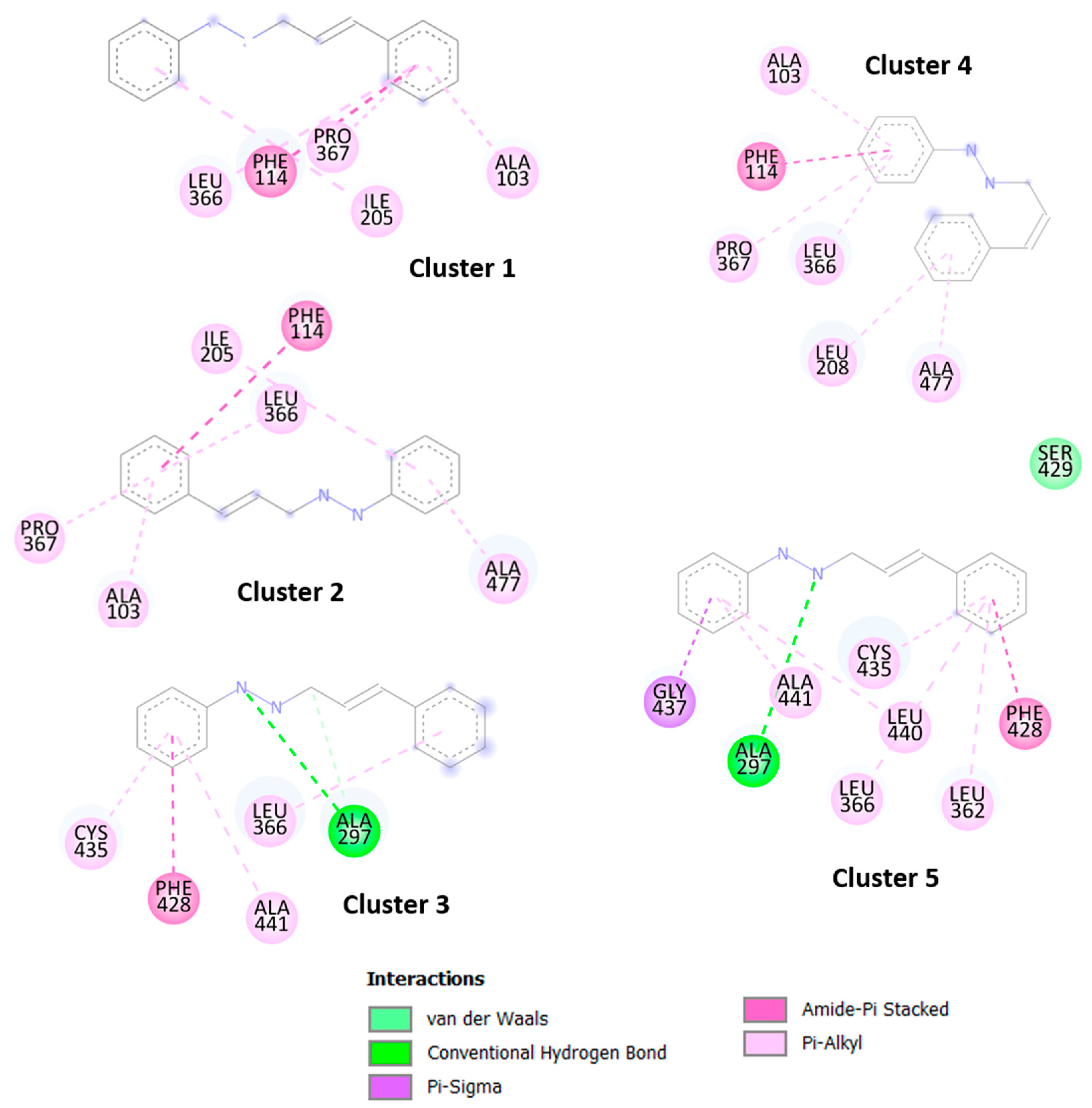
The results obtained by molecular docking protocols with SwissDock for the first cluster are shown in
Table 1 and
Figure 2 and
Figure 3. Predicted binding sites were clustered in 57 clusters with populations of 4–16 members. The cluster rank was predicted by the full fitness energy of the members. The best full fitness corresponded to the first member of each cluster, which is related to a better affinity of the ligand towards the protein.
Geometries of predicted binding sites corresponding to clusters 1–5 ranged from −2270.35 and −2267.43 Kcal/mol of full fitness values, the interaction types present between CPH and the CP-450 chain were identified. The observed interactions between CPH and the amino acid residues consisted of several hydrophobic interactions, especially with the residues corresponding to the sequence from 208–367 residues of amino acids. The cavity of the binding site contains aliphatic and aromatic hydrophobic residues such as Ala447, Phe428, Cys435, Leu366, Pro367, Phe144, Leu208, and Ile205, with a distance of interaction between CFH and residues of 3.5–5.1 Å. Similar interactions were observed with binding sites of other clusters not shown in
Figure 2 and
Figure 3. These results indicate that the CHP interacts with the hydrophobic sites of the central cavity of the enzyme.
3.2. Molecular Docking Calculation Conducted with CPH/NO
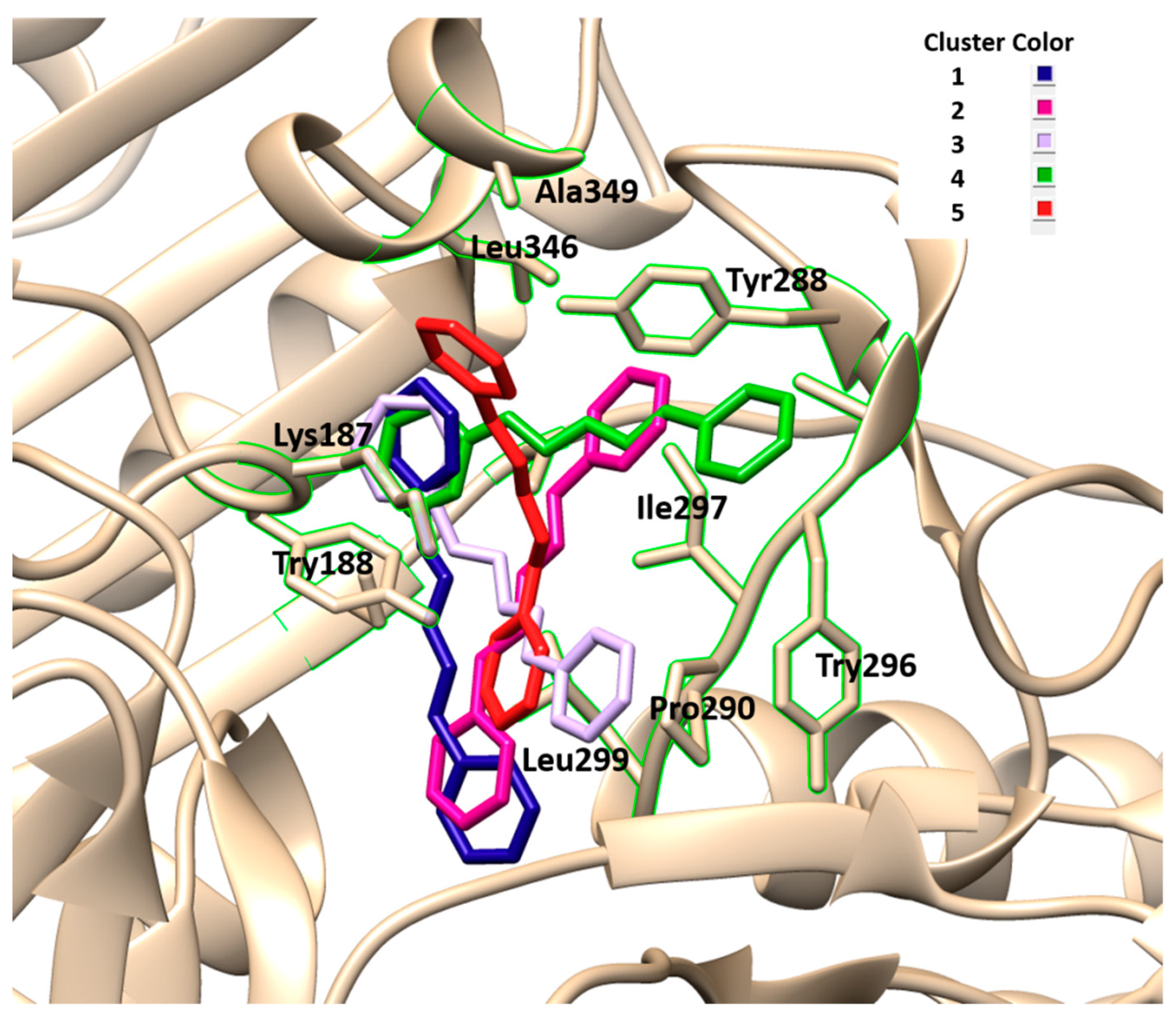
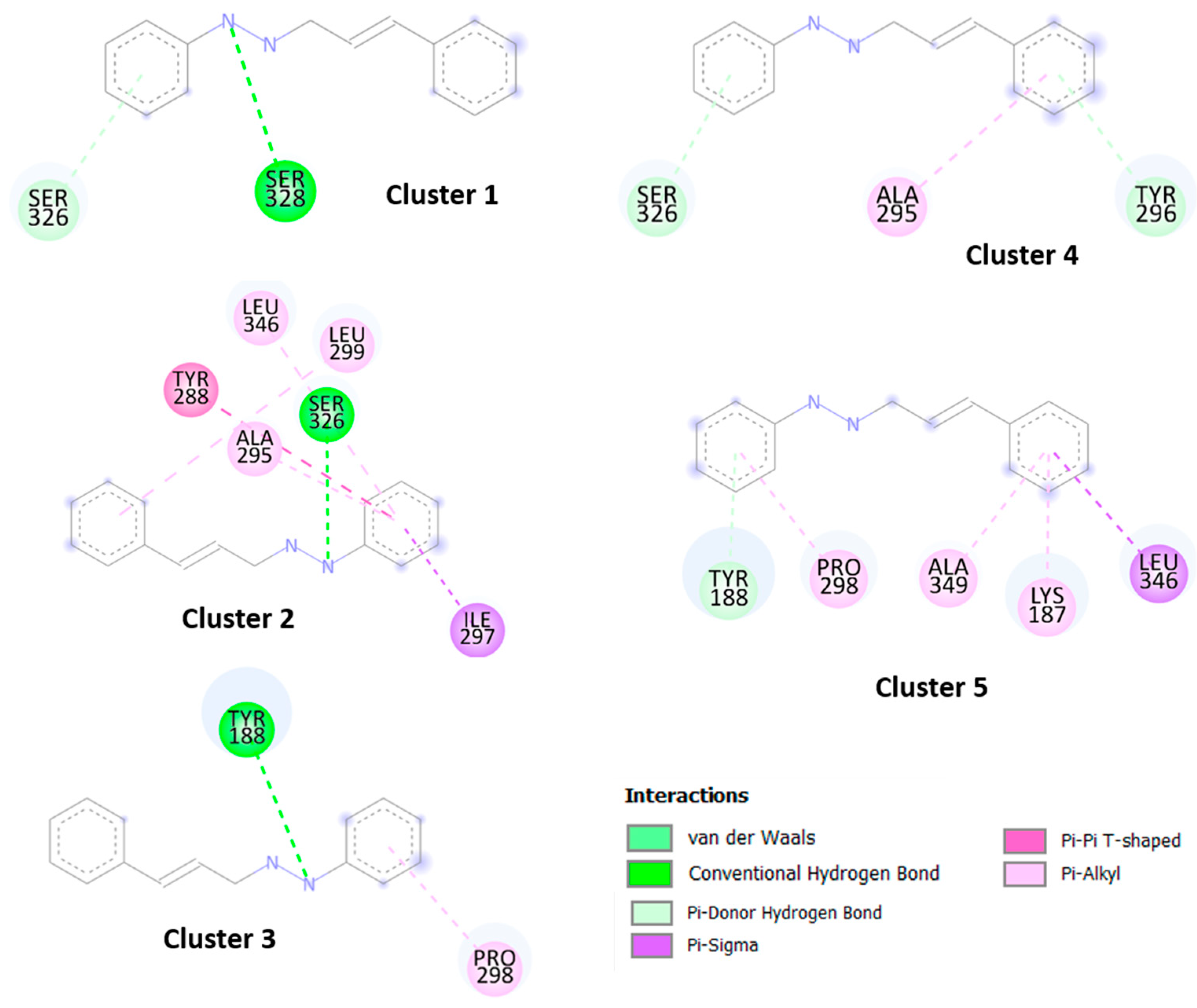
The results obtained by molecular docking protocols with SwissDock for the first cluster are shown in
Table 2 and
Figure 4 and
Figure 5. Predicted binding sites were clustered in 52 clusters with populations of 1–12 members. The cluster rank was predicted by the full fitness energy of the members. The best full fitness corresponded to the first member of each cluster.
From geometries of predicted binding sites corresponding to clusters 1–5 ranging from −2379.45 and −2374.23 Kcal/mol of full fitness values, the interaction types present in between CPH and the NO chain were identified. The observed interactions between CPH and the amino acid residues consisted of several hydrophobic interactions, especially with the residues corresponding to the sequence from 208–367 residues of amino acid. The cavity of the binding site contains aliphatic and aromatic hydrophobic residues such as Lys187. Try188, Try288, Ala295, Try296, Pro298, Ser326, Leu346, Ala349, with an interaction distance between CFH and residues of 3.0–4.7 Å. Similar interactions were observed with binding sites of other clusters not shown in
Figure 3 and
Figure 4. These results indicate that the CHP interacts with the hydrophobic sites of the enzyme.
4. Conclusions
In silico calculation is a useful tool to study the binding modes of CFH with CP-450 and NO enzymes, employing the SwissDock server.
If we compare the total energies, it is observed that, in both cases, the values are of the same order, so the affinity of the binding would be equivalent in both cases, although a little higher in the case of the NO enzyme. These values are high, implying a high affinity for the enzymes.
The observed interactions between CFH and the residues consisted of hydrophobic interactions of an electrostatic nature.
Author Contributions
Conceptualization, C.A.F.; methodology, C.A.F.; formal analysis, C.O.; writ-ing—original draft preparation, C.O.; writing—review and editing, C.A.F.; supervision, C.A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Universidad Nacional del Litoral (CAI+D2020) and the Ministerio de Ciencia, Tecnología e Innovación Productiva de Santa Fe (IO2019), Santa Fe, Ar-gentina.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This research was supported by Universidad Nacional del Litoral (CAI+D 2020) and the Ministerio de Ciencia, Tecnología e Innovación Productiva de Santa Fe (IO2019), Santa Fe, Argentina.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kareem, H.S.; Ariffin, A.; Nordin, N.; Heidelberg, T.; Abdul-Aziz, A.; Kong, K.W.; Yehye, W.A. Correlation of antioxidant activities with theoretical studies for new hydrazone compounds bearing a 3,4,5-trimethoxy benzyl moiety. Eur. J. Med. Chem. 2015, 103, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.G. Studies on the design, synthesis and application of active molecules with potential properties antioxidants and/or chemosensors. Unpublished.
- Chaudhary, K.K.; Mishra, N. A review on molecular docking: Novel tool for drug discovery. JSM Chem. 2016, 3, 1029. [Google Scholar]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The performance of current methods in ligand-protein docking. Curr. Sci. 2002, 83, 845–855. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Propka. Available online: http://server.poissonboltzmann.org/pdb2pqr (accessed on 20 August 2021).
- Mol Probity. Available online: http://molprobity.biochem.duke.edu/index.php (accessed on 20 August 2021).
- SwissDock Server. Available online: http://www.swissdock.ch/ (accessed on 22 August 2021).
- Discovery Studio Visualizer, v20.1.0.19259. 2009. Available online: https://discover.3ds.com/discovery-studio-visualizer/ (accessed on 21 August 2021).
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}