
A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles †


Abstract
:1. Introduction
2. Materials and Methods
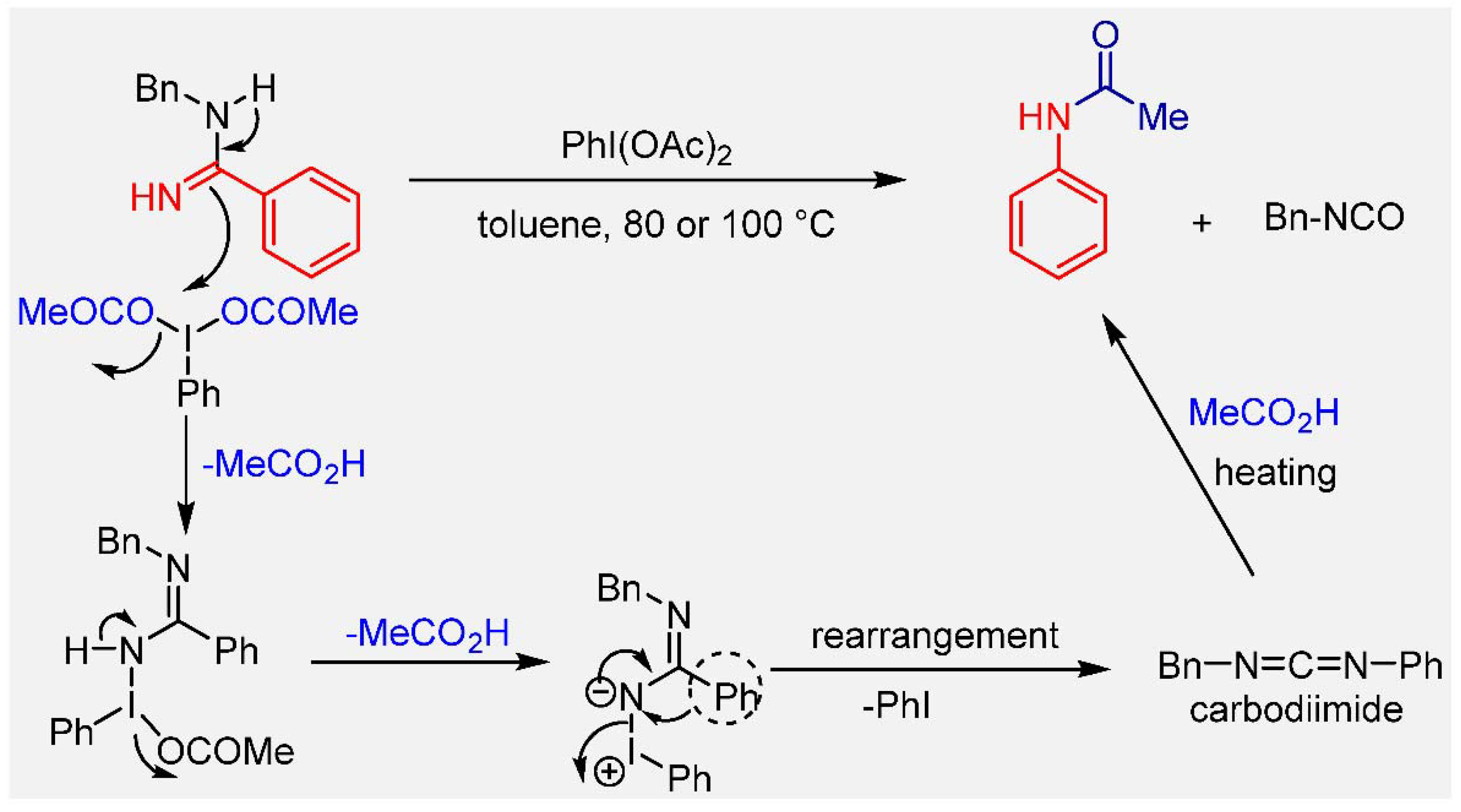
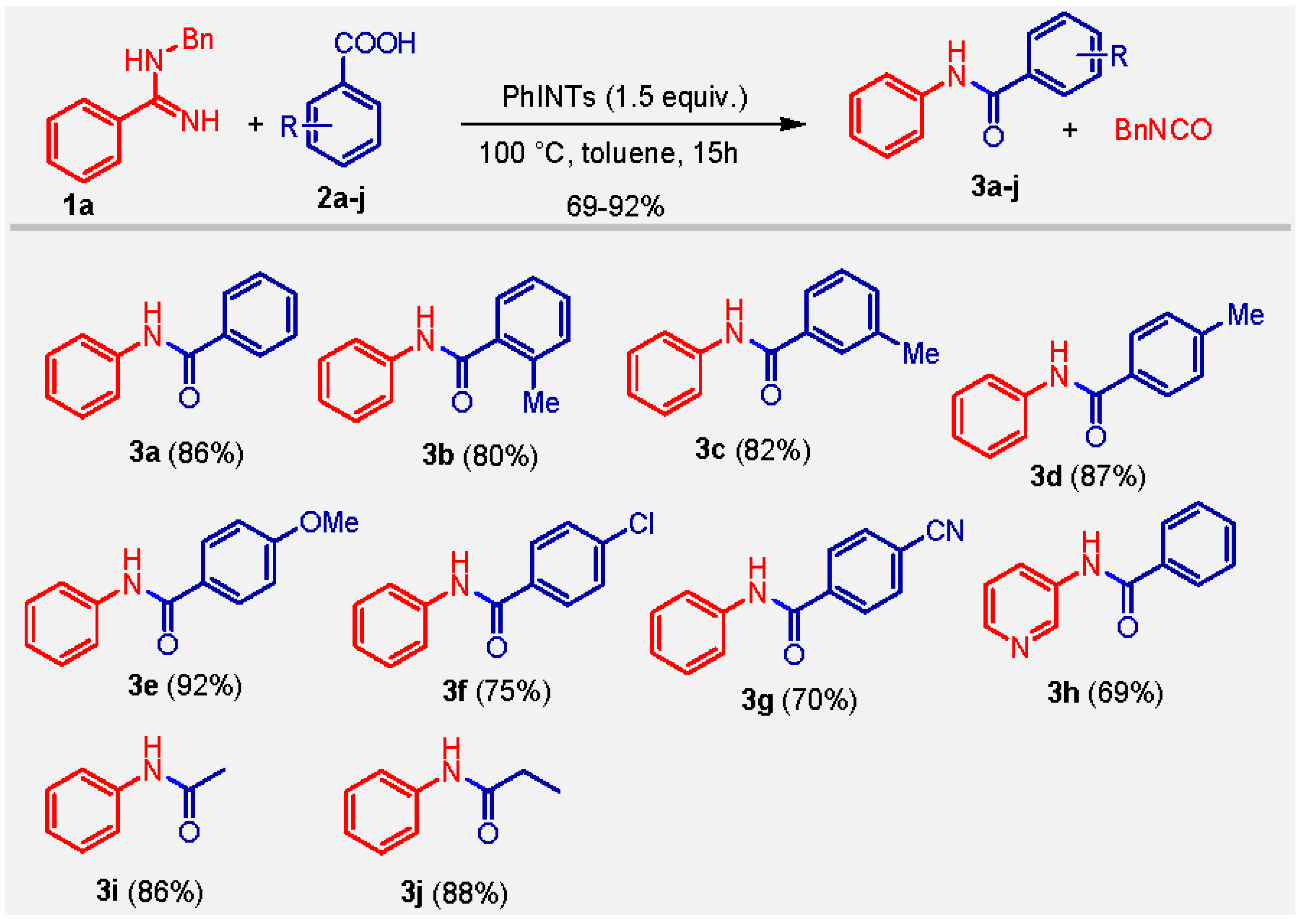
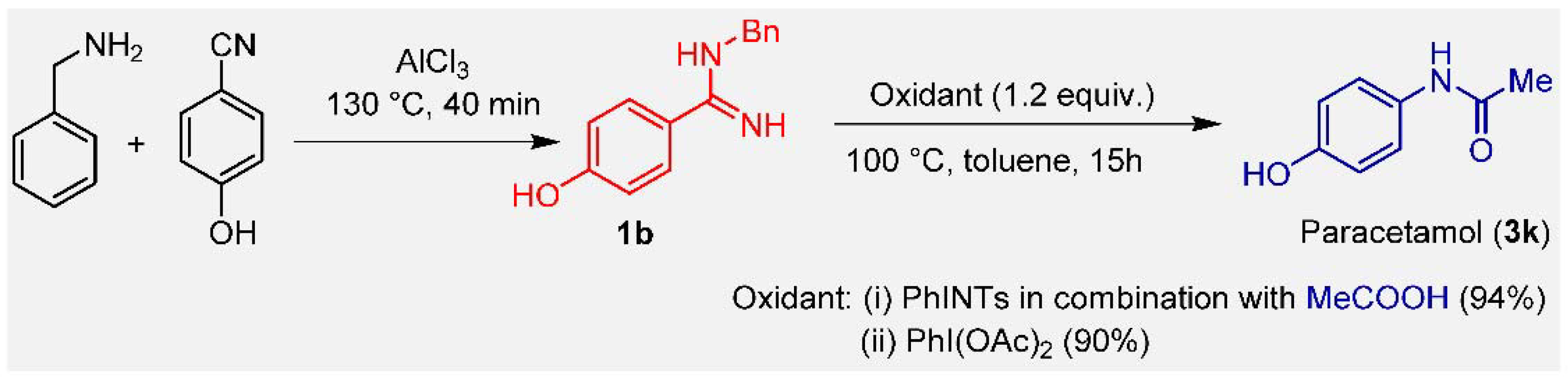
3. Results and Discussion
4. Conclusions
5. Experimental
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Humphrey, J.M.; Chamberlin, A.R. Chemical synthesis of natural product peptides: Coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 1997, 97, 2243–2266. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Sullivan, J.E.; Farrar, H.C. Fever and antipyretic use in children. Pediatrics 2011, 127, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Freund, Y.R.; Berry, P.; Ciaravino, V.; Erve, J.C.L.; Rosenthal, P.J.; et al. Benzoxaborole antimalarial agents. Part 5. Lead optimization of novel amide pyrazinyloxy benzoxaboroles and identification of a preclinical candidate. Med. Chem. 2017, 60, 5889–5908. [Google Scholar] [CrossRef] [PubMed]
- Walesa, K.G.; Stec, A.P. The synthesis and properties of N-substituted amides of 1-(5-methylthio-1,2,4-triazol-3-yl)-cyclohexane-2-carboxylic acid. Med. Acad. Lub. 2003, 9, 118–125. [Google Scholar]
- Warnecke, A.; Fichtner, I.; Sab, G.; Kratz, F. Synthesis, cleavage profile, and antitumor efficacy of an albumin-binding prodrug of methotrexate that is cleaved by plasmin and cathepsin B. Arch. Pharm. Chem. Life Sci. 2007, 340, 389–395. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Chen, Y.; Pienkowski, E.W.; Ju, J.; Lin, S.; Rajski, S.R.; Shen, B.J. Characterization of FdmV as an amide synthetase for fredericamycin A biosynthesis in Streptomyces griseus ATCC 43944. Biol. Chem. 2010, 285, 38853–38860. [Google Scholar] [CrossRef] [Green Version]
- Lakouraj, M.; Mokhtary, M.J. Synthesis of polyamides from p-Xylylene glycol and dinitriles. Polym. Res. 2009, 16, 681–686. [Google Scholar] [CrossRef]
- Bailey, P.D.; Mills, T.J.; Pettecrew, R.; Price, R.A. Comprehensive Organic Functional Group Transformations II: Carbon with No Attached Heteroatoms; Katrizky, A.R., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2005; Volume 5, p. 201. [Google Scholar]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Charville, H.; Jackson, D.; Hodges, G.; Whiting, A. The thermal and boron-catalysed direct amide formation reactions: Mechanistically understudied yet important processes. Chem. Commun. 2010, 46, 1813–1823. [Google Scholar] [CrossRef]
- Comerford, J.W.; Farmer, T.J.; MacQuarrie, D.J.; Breeden, S.W.; Clark, J.H. Mesoporous Structured Silica—An improved catalyst for direct amide synthesis and its application to continuous flow processing. Arkivoc 2012, 2012, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.L.; Williams, J.M.J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405–3415. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Buchwald, S.L. Pd-catalyzed intermolecular amidation of aryl halides: The discovery that xantphos can be trans-chelating in a palladium complex. Am. Chem. Soc. 2002, 124, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P. Recent advances in the synthesis of amides via oxime rearrangements and its applications. Curr. Org. Synth. 2018, 15, 666–706. [Google Scholar] [CrossRef]
- Wang, X.; Yang, P.; Hu, B.; Zhang, Q.; Li, D.J. Hypervalent iodine reagents promote a facile and efficient transformation of primary amides to secondary amides. Org. Chem. 2021, 86, 2820–2826. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Rose, H.L. Oxidative rearrangement and cyclisation of N-substituted amidines using iodine (III) reagents and the influence of leaving group on mode of reaction. Chem. Soc. Perkin Trans. 1 1997, 2319–2327. [Google Scholar] [CrossRef]
- Debnath, P.; Baeten, M.; LefÀvre, N.; Daele, S.V.; Maes, B.U.W. Synthesis of Secondary Amides from N-Substituted Amidines by Tandem Oxidative Rearrangement and Isocyanate Elimination. Adv. Synth. Catal. 2015, 357, 197–209. [Google Scholar] [CrossRef]
- Noe, M.; Perosa, A.; Selva, M. A flexible Pinner preparation of orthoesters: The model case of trimethylorthobenzoate. Green Chem. 2013, 15, 2252–2260. [Google Scholar] [CrossRef] [Green Version]
- Caskey, D.C.; Chapman, D.W. Process for Preparing p-Aminophenol and Alkyl Substituted p-Aminophenol. U.S. Patent 4,571,437, 18 February 1986. [Google Scholar]
- Rode, C.V.; Vaidya, M.J.; Chaudhari, R.V. Synthesis of p-aminophenol by catalytic hydrogenation of nitrobenzene. Organic process research & development. Org. Process Res. Dev. 1999, 3, 465–470. [Google Scholar]
- Joncour, R.; Duguet, N.; Metay, E.; Ferreira, A.; Lemaire, M. Amidation of phenol derivatives: A direct synthesis of paracetamol (acetaminophen) from hydroquinone. Green Chem. 2014, 16, 2997–3002. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Y.; Wang, Y.; Xue, W.; Zhao, X.J. Synthesis of p-aminophenol from the hydrogenation of nitrobenzene over metal–solid acid bifunctional catalyst. Chem. Technol. Biotechnol. 2008, 83, 1466–1471. [Google Scholar] [CrossRef]
- Liu, P.; Hu, Y.; Ni, M.; You, K.; Luo, H. Liquid phase hydrogenation of nitrobenzene to para-aminophenol over Pt/ZrO 2 catalyst and SO42−/ZrO2–Al2O3 solid acid. Catal. Lett. 2010, 140, 65–68. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}

| Entry | Oxidant (1.5 eq) | Solvent (1 mL) | Additives (1.1 eq) | Yield (%) 3a/4a |
|---|---|---|---|---|
| 1 | PhI(OAc)2 | Toluene | - | 48/40 |
| 2 | PhI(OCOCF3) | Toluene | - | 0/0 |
| 3 | PhINTs | Toluene | - | 86/0 |
| 4 | PhINTs | THF | - | 74/0 |
| 5 | PhINTs | DMF | - | 35/0 |
| 6 | PhINTs | o-Xylene | - | 68/0 |
| 7 | PhINTs | Toluene | Et3N | 77/0 |
| 8 | PhINTs | Toluene | AcOK | 58/34 |
| 9 | PhINTs | Toluene | Cs2CO3 | 42/0 |
| 10 | PhINTs | Toluene | Pyridine | 58/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debnath, P. A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles. Chem. Proc. 2022, 8, 27. https://doi.org/10.3390/ecsoc-25-11726
Debnath P. A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles. Chemistry Proceedings. 2022; 8(1):27. https://doi.org/10.3390/ecsoc-25-11726
Chicago/Turabian StyleDebnath, Pradip. 2022. "A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles" Chemistry Proceedings 8, no. 1: 27. https://doi.org/10.3390/ecsoc-25-11726
APA StyleDebnath, P. (2022). A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles. Chemistry Proceedings, 8(1), 27. https://doi.org/10.3390/ecsoc-25-11726