
Design, Development, and In Silico Study of Pyrazoline-Based Mycobactin Analogs as Anti-Tubercular Agents †



Abstract
:1. Introduction


2. Materials and Methods
2.1. Hardware and Software Employed
2.2. Docking Simulations
2.2.1. Protein Structure Preparation
2.2.2. Ligand Preparation
2.2.3. Protein–Ligand Docking Simulations
2.3. ADME Prediction
2.4. Toxicity Prediction
3. Results
3.1. Docking Simulation Studies
3.2. ADME Prediction
3.2.1. Results of Drug-Likeness, Bioavailability, Synthetic Feasibility, and Alerts for PAINS and Brenk Filters
3.2.2. In Silico Evaluation of Pharmacokinetic Compliance
3.3. Toxicity Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daniel, T.M.; Bates, J.H.; Downes, K.A. History of tuberculosis. Tuberc. Pathog. Prot. Control 1994, 13–24. [Google Scholar] [CrossRef]
- World Health Organization. Global Task Force on TB Impact Measurement. 2019. Available online: https://www.who.int/groups/global-task-force-on-tb-impact-measurement (accessed on 20 September 2021).
- World Health Organization. Tuberculosis; Key Facts. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 20 September 2021).
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The Mycobactin biosynthesis pathway: A prospective therapeutic target in the battle against tuberculosis. J. Med. Chem. 2020, 64, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.L. Breaking a pathogen’s iron will: Inhibiting siderophore production as an antimicrobial strategy. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2015, 1854, 1054–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Voss, J.J.; Rutter, K.; Schroeder, B.G.; Barry, C.E., III. Iron acquisition and metabolism by mycobacteria. J. Bacteriol. 1999, 181, 4443–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 396, 190. [Google Scholar] [CrossRef]
- de Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, S.; Guo, H.; Adamson, J.; Cisar, J.S.; Davis, T.D.; Chavadi, S.S.; Warren, J.D.; Quadri, L.E.N.; Tan, D.S.; Bishai, W.R. Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother. 2013, 57, 5138–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.V.; Puri, R.V.; Chauhan, P.; Kar, R.; Rohilla, A.; Khera, A.; Tyagi, A.K. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J. Infect. Dis. 2013, 208, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, B.P.; Nelson, K.M.; Aldrich, C.C. Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 2012, 12, 766–796. [Google Scholar] [CrossRef] [PubMed]
- Stirrett, K.L.; Ferreras, J.A.; Jayaprakash, V.; Sinha, B.N.; Ren, T.; Quadri, L.E.N. Small molecules with structural similarities to siderophores as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2008, 18, 2662–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreras, J.A.; Gupta, A.; Amin, N.D.; Basu, A.; Sinha, B.N.; Worgall, S.; Jayaprakash, V.; Quadri, L.E.N. Chemical scaffolds with structural similarities to siderophores of nonribosomal peptide–polyketide origin as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2011, 21, 6533–6537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes. BIOVIA Discovery Studio Visualizer; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Sl. No. | Code | R | R1 |
|---|---|---|---|
| 01 | GM01 |  | 2-CH3 |
| 02 | GM02 |  | 3-CH3 |
| 03 | GM03 |  | 4-CH3 |
| 04 | GM04 |  | 2-OCH3 |
| 05 | GM05 |  | 3-OCH3 |
| 06 | GM06 |  | 4-OCH3 |
| 07 | GM07 |  | 2-Cl |
| 08 | GM08 |  | 3-Cl |
| 09 | GM09 |  | 4-Cl |
| 10 | GM10 |  | 2-OH |
| 11 | GM11 |  | 3-OH |
| 12 | GM12 |  | 4-OH |
| Sl No. | Code | Dock Score | Inhibition Constant |
|---|---|---|---|
| 01 | GM01 | −9.23 | 171.39 nM |
| 02 | GM02 | −9.72 | 74.65 nM |
| 03 | GM03 | −9.64 | 85.71 nM |
| 04 | GM04 | −9.19 | 182.87 nM |
| 05 | GM05 | −9.41 | 126.09 nM |
| 06 | GM06 | −9.49 | 110.58 nM |
| 07 | GM07 | −9.32 | 148.14 nM |
| 08 | GM08 | −9.90 | 55.54 nM |
| 09 | GM09 | −9.83 | 62.70 nM |
| 10 | GM10 | −8.58 | 510.97 nM |
| 11 | GM11 | −9.04 | 235.3 nM |
| 12 | GM12 | −8.86 | 317.69 nM |
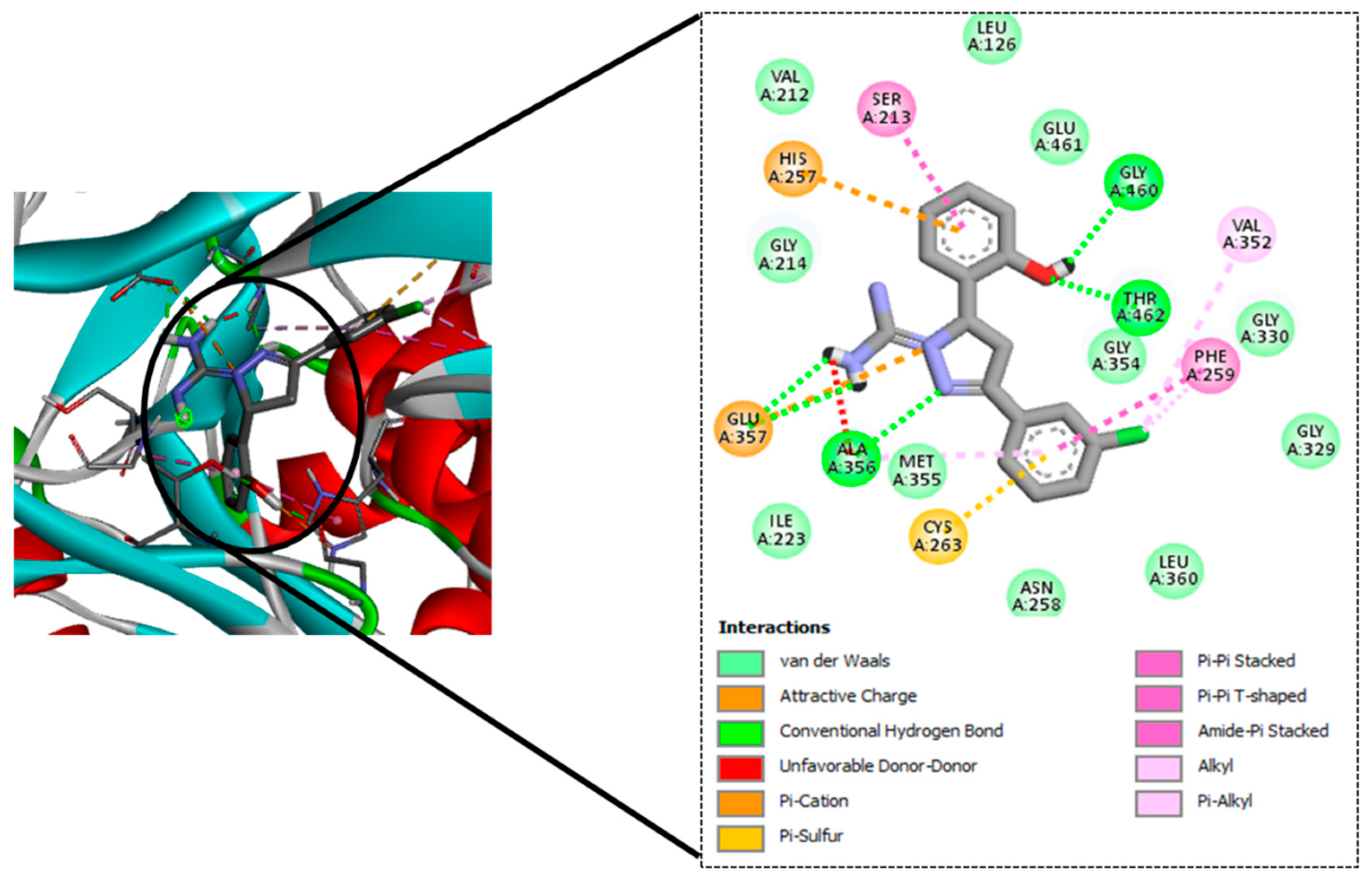
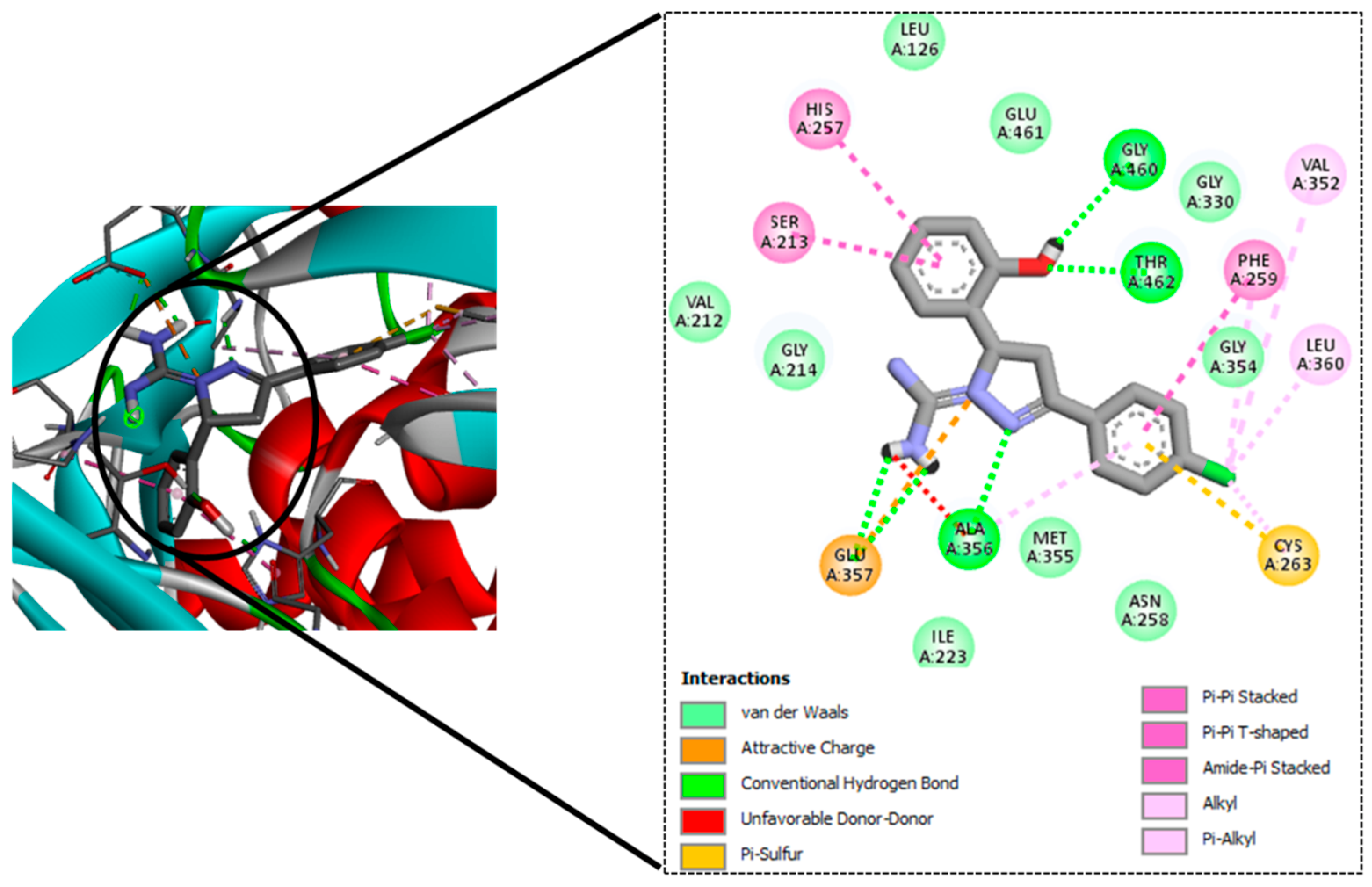
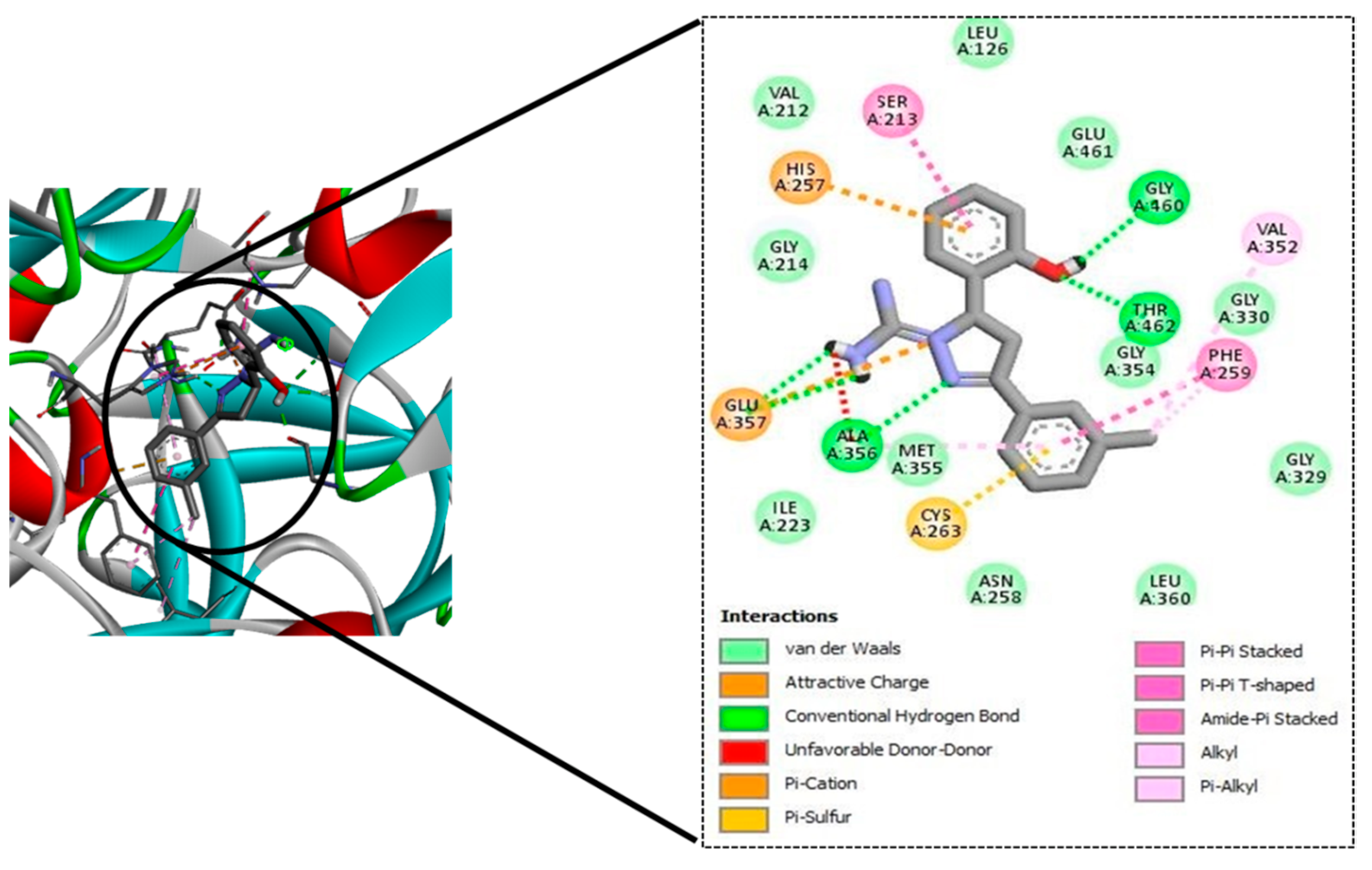
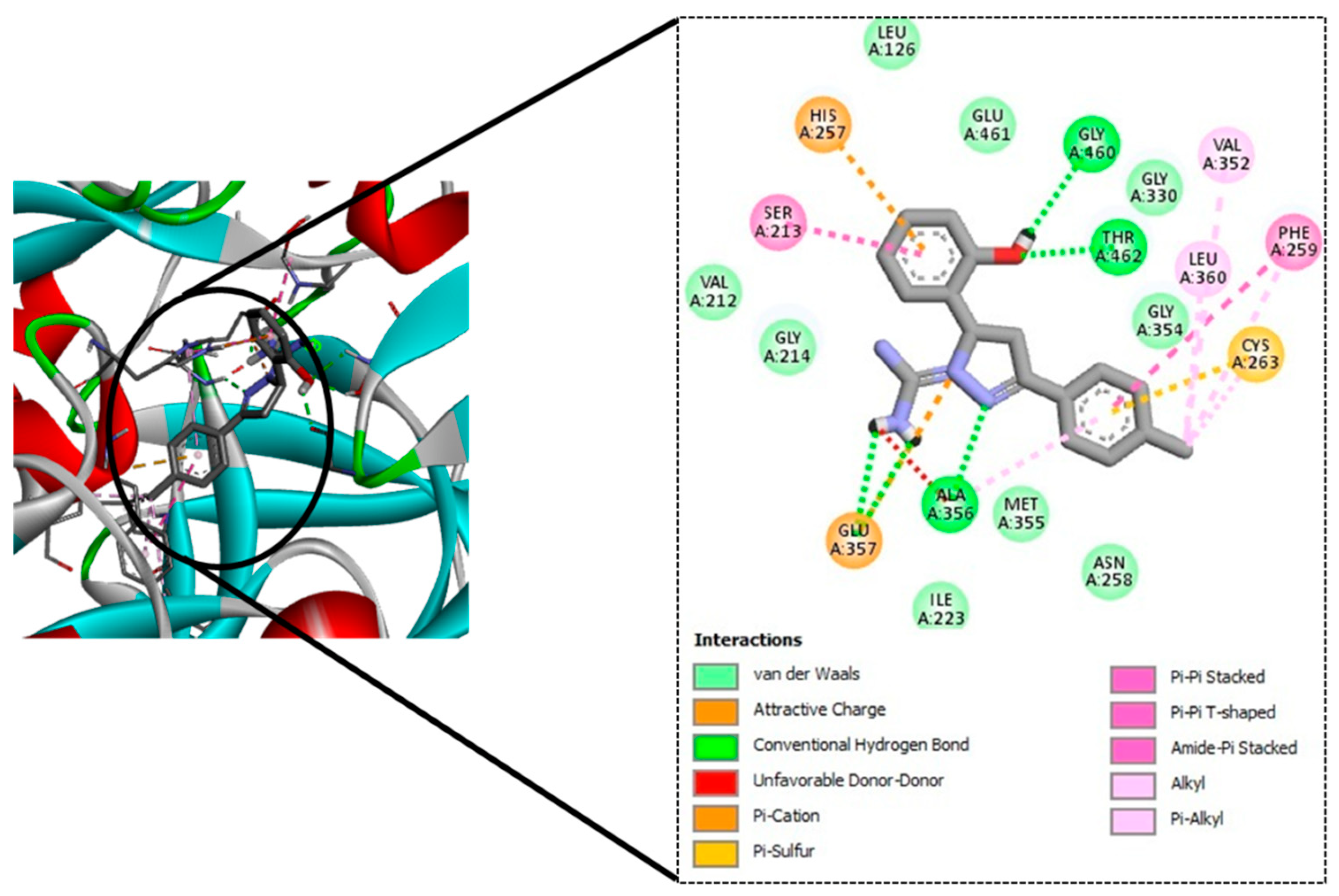
| Sl No. | Ligand Code | H-Bond Residues |
|---|---|---|
| 1. | GM08 | Gly460, Thr462, Ala356 |
| 2. | GM09 | Gly460, Thr462, Ala356 |
| 3. | GM02 | Gly460, Thr462, Ala356 |
| 4. | GM03 | Gly460, Thr462, Ala356 |
| Sl No. | Compound Code | Drug-Likeness Rules | Alerts | Lead-Likeness | Synthetic Accessibility | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipinski (Pfizer) | Ghose (Amgen) | Veber (GSK) | Egan (Pharmacia) | Muege (Bayer) | Bioavailability Score | PAINS | Brenk | ||||
| 1. | GM08 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 2 | Yes | 3.52 |
| 2. | GM09 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 2 | Yes | 3.52 |
| 3. | GM02 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 2 | Yes | 3.65 |
| 4. | GM03 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 2 | Yes | 3.63 |
| ADMET PROFILE | GM08 | GM09 | GM02 | GM03 | ||
| Physiochemical Parameters | Formula | C16H15ClN4O | C16H15ClN4O | C17H18N4O | C17H18N4O | |
| Molecular Weight | 314.77 g/mol | 314.77 g/mol | 294.35 g/mol | 294.35 g/mol | ||
| Mol. Refractivity | 95.36 | 95.36 | 95.31 | 95.31 | ||
| TPSA | 85.70 Å2 | 85.70 Å2 | 85.70 Å2 | 85.70 Å2 | ||
| Lipophilicity | ILOGP | 2.05 | 2.02 | 2.02 | 2.10 | |
| SILICOS-IT | 2.57 | 2.57 | 2.44 | 2.44 | ||
| Water Solubility | Log S (ESOL), Class | −3.57 | −3.57 | −3.28 | −3.28 | |
| Log S (Ali), Class | −3.93 | −3.93 | −3.66 | −3.66 | ||
| SILICOS-IT, Class | −4.64 | −4.64 | −4.42 | −4.42 | ||
| Pharmacokinetics | GI Absorption | High | High | High | High | |
| BBB Permeant | No | No | No | No | ||
| Log Kp (Skin Permeant) | −6.45 cm/s | −6.45 cm/s | −6.51 cm/s | −6.51 cm/s | ||
| CYP1A2 | Yes | Yes | No | No | ||
| CYP2C19 | No | No | No | No | ||
| CYP2C9 | No | No | No | No | ||
| CYP2D6 | No | No | No | No | ||
| CYP3A4 | No | No | No | No | ||
| Model Name | Units | GM08 | GM09 | GM02 | GM03 |
|---|---|---|---|---|---|
| AMES Toxicity | Yes/No | Yes | No | Yes | Yes |
| Max. Tolerated Dose (Human) | Log mg/kg/day | −0.127 | −0.177 | −0.15 | −0.199 |
| hERG I inhibitor | Yes/No | No | No | No | No |
| hERG II inhibitor | Yes/No | Yes | Yes | Yes | Yes |
| Oral Rat Chronic Toxicity (LD50) | Mol/kg | 3.272 | 3.273 | 3.257 | 3.258 |
| Oral Rat Chronic Toxicity | Log mg/kg_bw/day | 1.498 | 1.566 | 1.415 | 1.519 |
| Hepatotoxicity | Yes/No | No | No | No | No |
| Skin Sensitization | Yes/No | No | No | No | No |
| T. Pyriformis Toxicity | Log ug/L | 0.264 | 0.274 | 0.262 | 0.271 |
| Minnow Toxicity | Log mM | 1.143 | 1.396 | 1.361 | 1.614 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakshit, G.; Murtuja, S.; Jayaprakash, V. Design, Development, and In Silico Study of Pyrazoline-Based Mycobactin Analogs as Anti-Tubercular Agents. Chem. Proc. 2022, 8, 62. https://doi.org/10.3390/ecsoc-25-11767
Rakshit G, Murtuja S, Jayaprakash V. Design, Development, and In Silico Study of Pyrazoline-Based Mycobactin Analogs as Anti-Tubercular Agents. Chemistry Proceedings. 2022; 8(1):62. https://doi.org/10.3390/ecsoc-25-11767
Chicago/Turabian StyleRakshit, Gourav, Sheikh Murtuja, and Venkatesan Jayaprakash. 2022. "Design, Development, and In Silico Study of Pyrazoline-Based Mycobactin Analogs as Anti-Tubercular Agents" Chemistry Proceedings 8, no. 1: 62. https://doi.org/10.3390/ecsoc-25-11767
APA StyleRakshit, G., Murtuja, S., & Jayaprakash, V. (2022). Design, Development, and In Silico Study of Pyrazoline-Based Mycobactin Analogs as Anti-Tubercular Agents. Chemistry Proceedings, 8(1), 62. https://doi.org/10.3390/ecsoc-25-11767