
Genetic Monitoring of the Captive Population of the Critically Endangered Brazilian Merganser (Mergus octosetaceus)
 ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Samples
2.2. DNA Extraction
2.3. ddRAD Library Construction and Sequencing
2.4. Variant Calling and Filtering
2.5. Statistical Genetics Analyses
3. Results
3.1. Variant Calling from Sequencing Reads
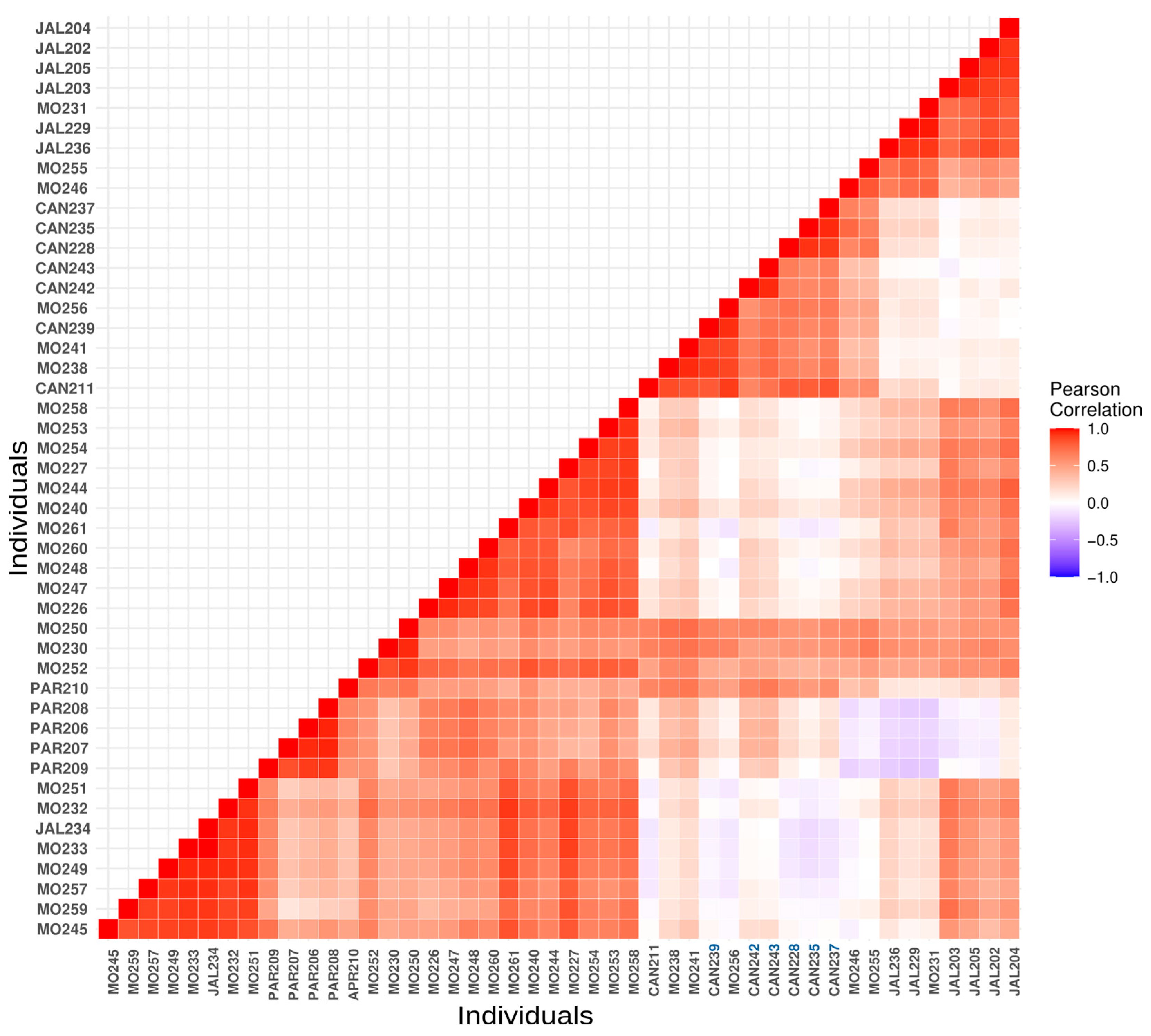
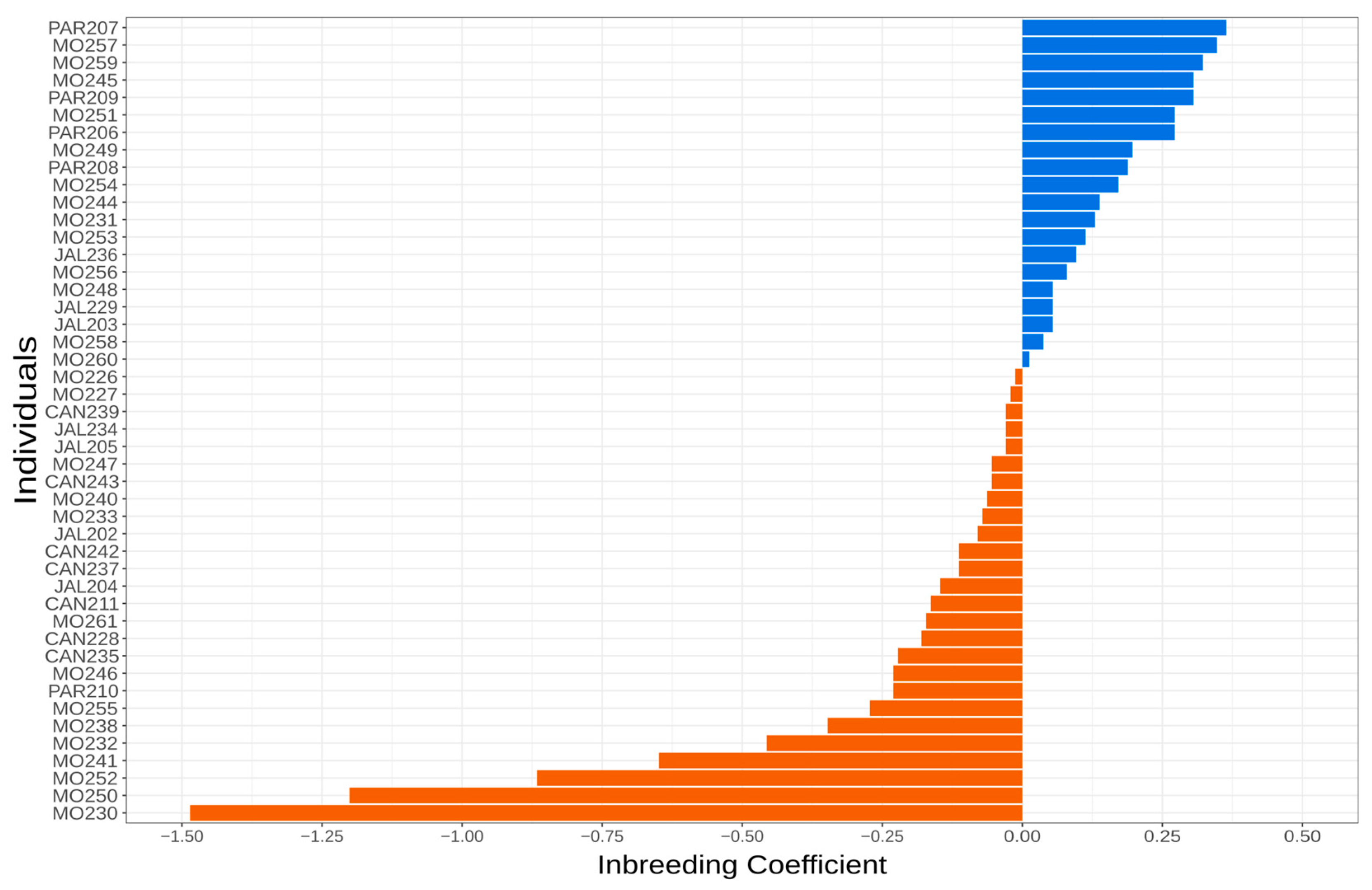
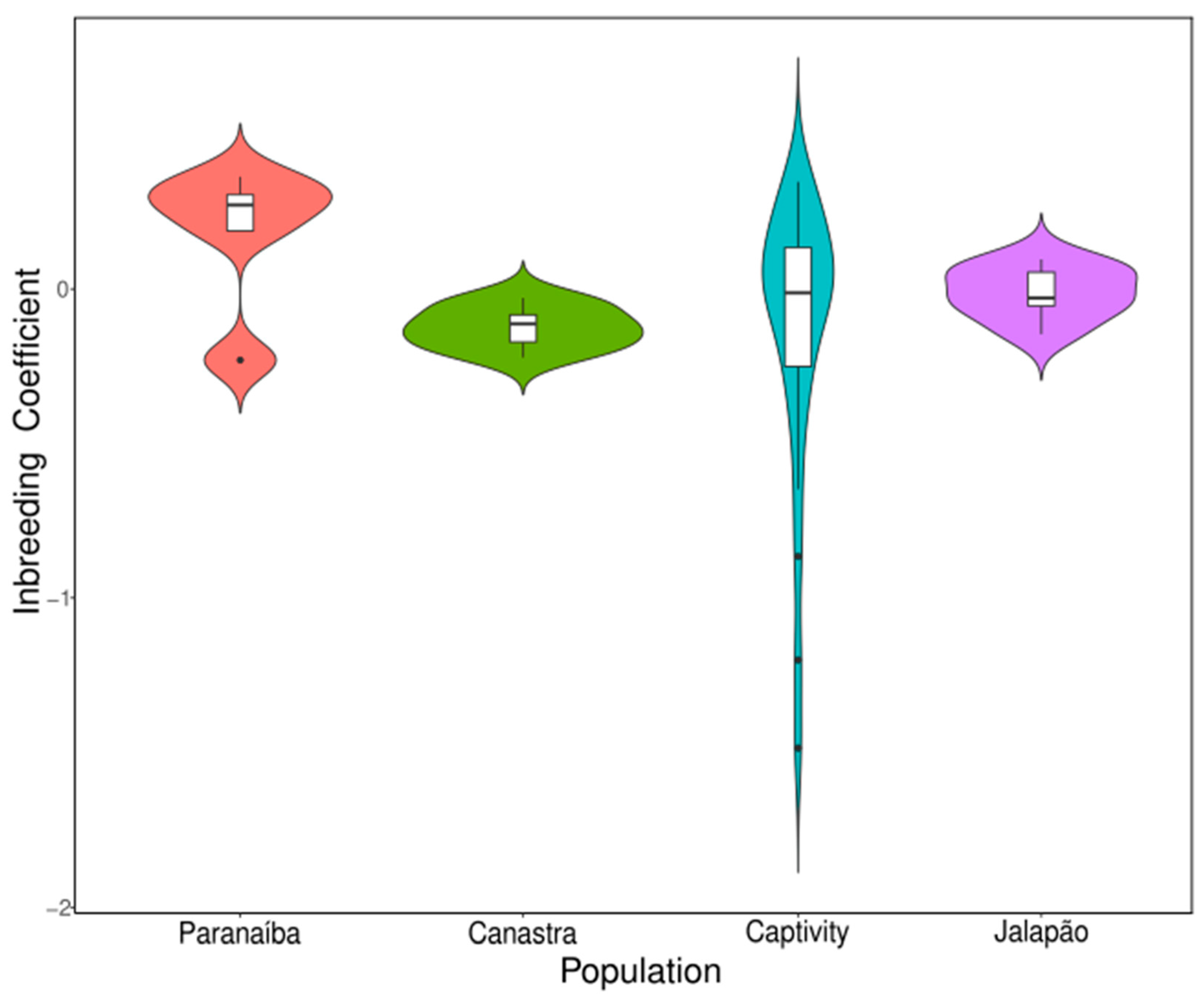
3.2. Kinship and Inbreeding Estimates
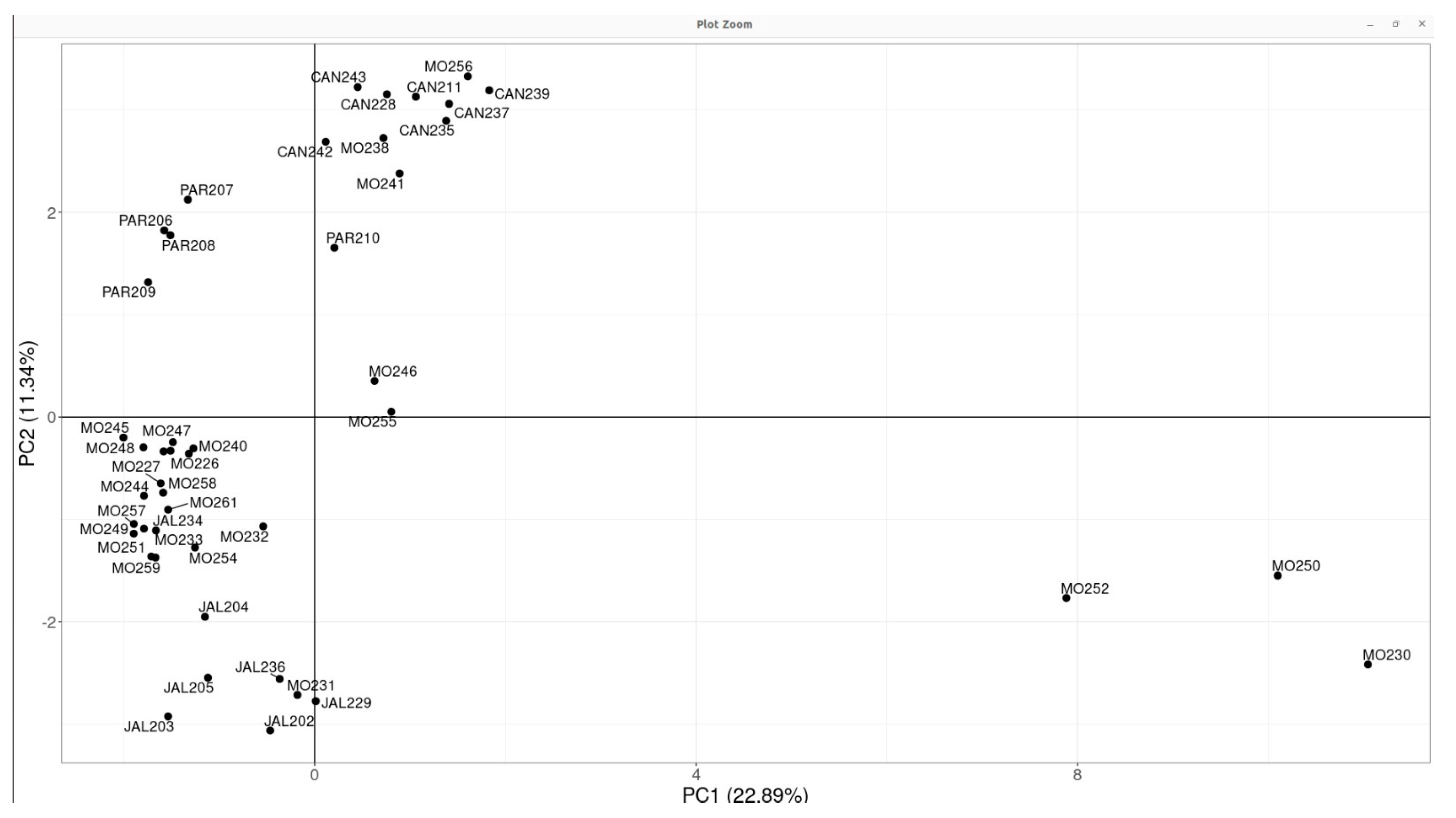
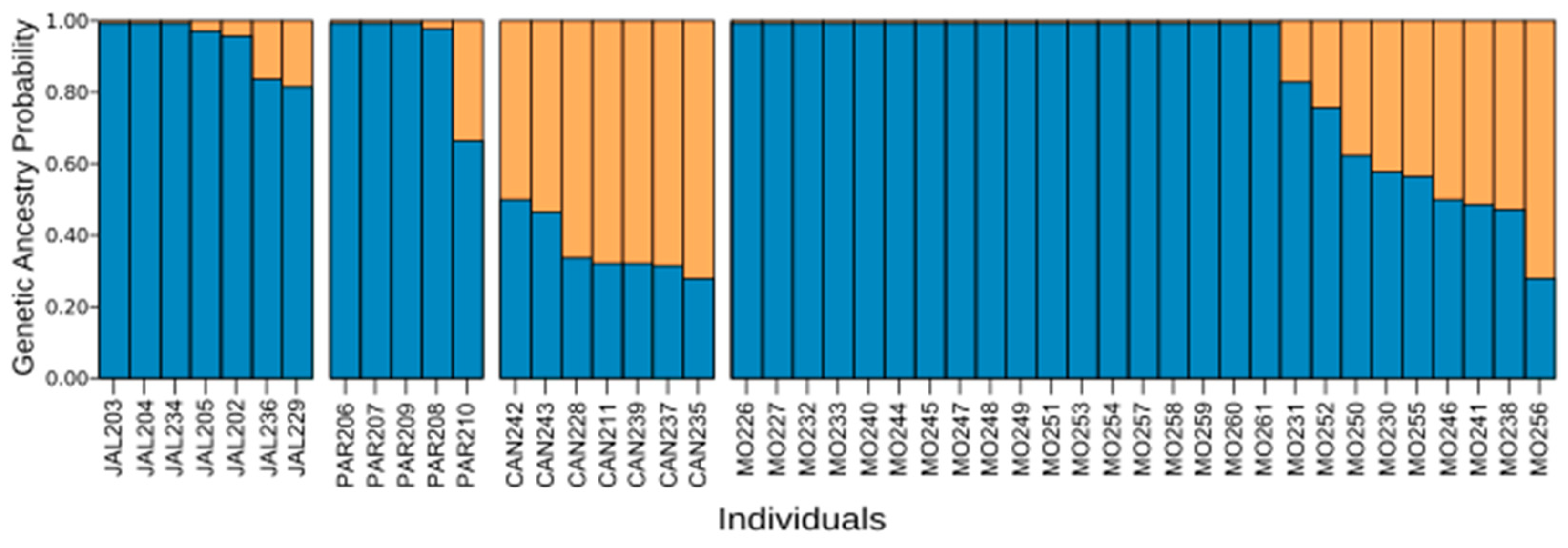
3.3. Captive Population Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collar, N.J.; Gonzaga, L.P.; Krabbe, N.; Madrono Nieto, A.; Naranjo, L.G.; Parker, T.A.; Wege, D.C. Threatened Birds of the Americas; IUCN and ICBP (International Council for Bird Preservation): Cambridge, UK, 1992; ISBN 978-1-56098-267-8. [Google Scholar]
- Antas, P. The Brazilian Merganser (Mergus octosetaceus), the Most Threathened Duck in South America. Gibier Faune Sauvag. 1996, 13, 799–800. [Google Scholar]
- IUCN. BirdLife International: The IUCN Red List of Threatened Species: Mergus octosetaceus. 2019. Available online: https://www.iucnredlist.org/species/22680482/143756439 (accessed on 7 November 2023).
- Yamashita, C.; Valle, M.D.P. Ocorrência de duas aves raras no Brasil Central: Mergus octosetaceus e Tigrisoma fasciatum. Ararajuba 1990, 1, 107–109. [Google Scholar]
- Benstead, P.J.; Hearn, R.D.; Nedelcoff, A.R.S. A recent sighting of Brazilian Merganser Mergus octosetaceus in Misiones province, Argentina. Cotinga 1994, 2, 35–36. [Google Scholar]
- Hearn, R. The current status of the Brazilian Merganser (Mergus octosetaceus) in Argentina. TWSG News 1994, 5, 14–15. [Google Scholar]
- Fragano, F.; Clay, R. Biodiversity status of the interior Atlantic forest of Paraguay. In The Atlantic Forest of South America: Biodiversity Status, Threats, and Outlook; Galindo-Leal, C., Câmara, I.G., Eds.; Island Press: Washington, DC, USA, 2003; p. 488. ISBN 1-55963-988-1. [Google Scholar]
- Rose, P. Evidence for Aviculture: Identifying Research Needs to Advance the Role of Ex Situ Bird Populations in Conservation Initiatives and Collection Planning. Birds 2021, 2, 77–95. [Google Scholar] [CrossRef]
- Vilaça, S.T.; Redondo, R.A.F.; Lins, L.V.; Santos, F.R. Remaining genetic diversity in Brazilian Merganser (Mergus octosetaceus). Conserv. Genet. 2012, 13, 293–298. [Google Scholar] [CrossRef]
- Maia, T.A.; Vilaça, S.T.; Silva, L.R.D.; Santos, F.R.; Dantas, G.P.D.M. DNA sampling from eggshells and microsatellite genotyping in rare tropical birds: Case study on Brazilian Merganser. Genet. Mol. Biol. 2017, 40, 808–812. [Google Scholar] [CrossRef]
- Faux, P.; Oliveira, J.C.P.; Campos, D.P.; Dantas, G.P.M.; Maia, T.A.; Dergan, C.G.; Cassemiro, P.M.; Hajdu, G.L.; Santos-Júnior, J.E.; Santos, F.R. Fast genomic analysis of aquatic bird populations from short single-end reads considering sex-related pitfalls. Ecol. Inform. 2020, 56, 101058. [Google Scholar] [CrossRef]
- Maia, T.A.; Campos, D.P.; Silva, L.R.; Lins, L.V.; Ribeiro, F.; Sebaio, F.; Rodrigues, F.R.; Dantas, G.P.M. Evidence of strong population bottleneck in genetics of endangered Brazilian Merganser (Mergus octosetaceus). J. Ornithol. 2020, 161, 521–527. [Google Scholar] [CrossRef]
- Campos, D.P.; Granger-Neto, H.P.; Santos-Júnior, J.E.; Faux, P.; Santos, F.R. Population genomics of the critically endangered Brazilian merganser. Animals 2023, 13, 3759. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetic adaptation to captivity in species conservation programs. Mol. Ecol. 2008, 17, 325–333. [Google Scholar] [CrossRef]
- Toone, W.D.; Wallace, M.P. The Extinction in the Wild and Reintroduction of the California Condor (Gymnogyps californianus). In Creative Conservation: Interactive Management of Wild and Captive Animals; Olney, P.J.S., Mace, G.M., Feistner, A.T.C., Eds.; Springer: Dordrecht, The Netherlands, 1994; pp. 411–419. ISBN 978-94-011-0721-1. [Google Scholar]
- van Dierendonck, M.C.; de Vries, M.F.W. Ungulate Reintroductions: Experiences with the Takhi or Przewalski Horse (Equus ferus przewalskii) in Mongolia. Conserv. Biol. 1996, 10, 728–740. [Google Scholar] [CrossRef]
- Fischer, J.; Lindenmayer, D.B. An assessment of the published results of animal relocations. Biol. Conserv. 2000, 96, 1–11. [Google Scholar] [CrossRef]
- Williams, S.E.; Hoffman, E.A. Minimizing genetic adaptation in captive breeding programs: A review. Biol. Conserv. 2009, 142, 2388–2400. [Google Scholar] [CrossRef]
- Waples, R.S.; Drake, J. Risk/Benefit Considerations for Marine Stock Enhancement: A Pacific Salmon Perspective. In Stock Enhancement and Sea Ranching; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 260–306. ISBN 978-0-470-75132-9. [Google Scholar]
- Melampy, M.N.; Howe, H.F. Sex Ratio in the Tropical Tree Triplaris americana (Polygonaceae). Evolution 1977, 31, 867–872. [Google Scholar] [CrossRef]
- Crow, J.F.; Denniston, C. Inbreeding and Variance Effective Population Numbers. Evolution 1988, 42, 482–495. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Torati, L.S.; Taggart, J.B.; Varela, E.S.; Araripe, J.; Wehner, S.; Migaud, H. Genetic diversity and structure in Arapaima gigas populations from Amazon and Araguaia-Tocantins river basins. BMC Genet. 2019, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.M.; Williams, C.K.; Lavretsky, P.; Howell, D.L.; Fuller, J.C. Mallard–Black Duck Hybridization and Population Genetic Structure in North Carolina. J. Wildl. Manag. 2021, 85, 1243–1255. [Google Scholar] [CrossRef]
- Diaz-Martin, Z.; Fant, J.; Havens, K.; Cinea, W.; Tucker Lima, J.M.; Griffith, M.P. Current management practices do not adequately safeguard endangered plant species in conservation collections. Biol. Conserv. 2023, 280, 109955. [Google Scholar] [CrossRef]
- Ba, H.; Jia, B.; Wang, G.; Yang, Y.; Kedem, G.; Li, C. Genome-Wide SNP Discovery and Analysis of Genetic Diversity in Farmed Sika Deer (Cervus nippon) in Northeast China Using Double-Digest Restriction Site-Associated DNA Sequencing. G3 2017, 7, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Çilingir, F.G.; Hansen, D.; Ozgul, A.; Grossen, C. Design of SNP markers for Aldabra giant tortoises using low coverage ddRAD-Seq. Conserv. Genet. Resour. 2021, 13, 409–412. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 2001; Volume 1. [Google Scholar]
- Thrasher, D.J.; Butcher, B.G.; Campagna, L.; Webster, M.S.; Lovette, I.J. Double-Digest RAD sequencing outperforms microsatellite loci at assigning paternity and estimating relatedness: A proof of concept in a highly promiscuous bird. Mol. Ecol. Resour. 2018, 18, 953–965. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Knaus, B.J.; Grünwald, N.J. vcfr: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 7 November 2023).
- Slowikowski, K.; Schep, A.; Hughes, S.; Dang, T.K.; Lukauskas, S.; Irisson, J.-O.; Kamvar, Z.N.; Ryan, T.; Christophe, D.; Hiroaki, Y.; et al. Ggrepel: Automatically Position Non-Overlapping Text Labels with “ggplot2”. 2023. Available online: https://rdrr.io/cran/ggrepel (accessed on 12 March 2024).
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Soft. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickhaml, H.; Henry, L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. Available online: https://ggplot2.tidyverse.org/reference/ggplot2-package.html (accessed on 18 November 2023).
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Waples, R.S.; Antao, T.; Luikart, G. Effects of Overlapping Generations on Linkage Disequilibrium Estimates of Effective Population Size. Genetics 2014, 197, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.; Dugger, B.; Cunha, H.; Lamas, I.; Goerck, J.; Lins, L.; Silveira, L.; Andrade, R.; Bruno, S.; Rigueira, S.; et al. Plano de Ação Para a Conservação Do Pato-Mergulhão (Mergus octosetaceus); IBAMA: Alta Floresta, Brazil, 2006; ISBN 978-85-7300-226-3. [Google Scholar]
- Brandt, A.; Tran Van, P.; Bluhm, C.; Anselmetti, Y.; Dumas, Z.; Figuet, E.; François, C.M.; Galtier, N.; Heimburger, B.; Jaron, K.S.; et al. Haplotype divergence supports long-term asexuality in the oribatid mite Oppiella nova. Proc. Natl. Acad. Sci. USA 2021, 118, e2101485118. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.; Krzywinski, M.; Altman, N. Principal component analysis. Nat. Methods 2017, 14, 641–642. [Google Scholar] [CrossRef]
- Frankham, R. Effective population size/adult population size ratios in wildlife: A review. Genet. Res. 1995, 66, 95–107. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Elam, D.R. Population Genetic Consequences of Small Population Size: Implications for Plant Conservation. Annu. Rev. Ecol. Syst. 1993, 24, 217–242. [Google Scholar] [CrossRef]
- Nadeau, N.J.; Mundy, N.I.; Gourichon, D.; Minvielle, F. Association of a single-nucleotide substitution in TYRP1 with roux in Japanese quail (Coturnix japonica). Anim. Genet. 2007, 38, 609–613. [Google Scholar] [CrossRef]
- Grouw, H. What Colour Is That Bird? The causes and recognition of common colour aberrations in birds. Br. Birds 2013, 106, 17–29. [Google Scholar]
- Díez-del-Molino, D.; von Seth, J.; Gyllenstrand, N.; Widemo, F.; Liljebäck, N.; Svensson, M.; Sjögren-Gulve, P.; Dalén, L. Population genomics reveals lack of greater white-fronted introgression into the Swedish lesser white-fronted goose. Sci. Rep. 2020, 10, 18347. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Jalapão | Paranaíba | Canastra | Captive-Bred | |
|---|---|---|---|---|
| Jalapão | - | - | - | - |
| Paranaíba | 0.2450 | - | - | - |
| Canastra | 0.1934 | 0.1126 | - | - |
| Captive-bred | 0.0570 | 0.0750 | 0.1018 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, D.P.; Granger-Neto, H.P.; Santos-Júnior, J.E.; Buzatti, R.S.O.; Santos, F.R. Genetic Monitoring of the Captive Population of the Critically Endangered Brazilian Merganser (Mergus octosetaceus). Birds 2024, 5, 190-201. https://doi.org/10.3390/birds5010013
Campos DP, Granger-Neto HP, Santos-Júnior JE, Buzatti RSO, Santos FR. Genetic Monitoring of the Captive Population of the Critically Endangered Brazilian Merganser (Mergus octosetaceus). Birds. 2024; 5(1):190-201. https://doi.org/10.3390/birds5010013
Chicago/Turabian StyleCampos, Davidson P., Henry P. Granger-Neto, José E. Santos-Júnior, Renata S. O. Buzatti, and Fabrício R. Santos. 2024. "Genetic Monitoring of the Captive Population of the Critically Endangered Brazilian Merganser (Mergus octosetaceus)" Birds 5, no. 1: 190-201. https://doi.org/10.3390/birds5010013
APA StyleCampos, D. P., Granger-Neto, H. P., Santos-Júnior, J. E., Buzatti, R. S. O., & Santos, F. R. (2024). Genetic Monitoring of the Captive Population of the Critically Endangered Brazilian Merganser (Mergus octosetaceus). Birds, 5(1), 190-201. https://doi.org/10.3390/birds5010013