
Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis

{kind=link}
Abstract
:1. AL Amyloidosis: A Plasma Cell Neoplasm with Long-Distance Effects
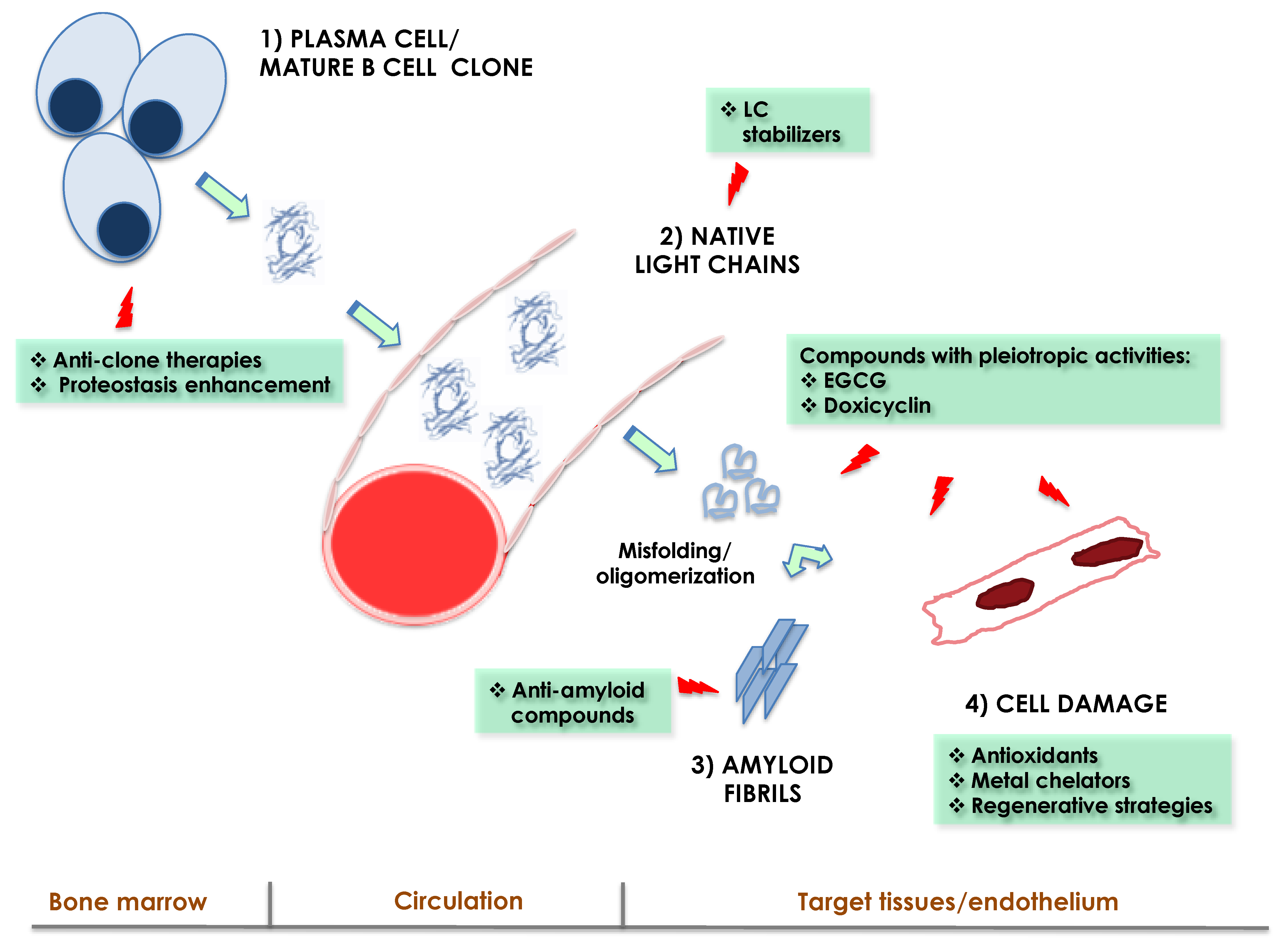
2. Damage Mechanisms in AL: Lessons from Clinical Observations, Diseased Tissues and Experimental Models
2.1. Two Players in Damage: Amyloid Fibrils and Precursor LCs
2.2. Soluble LC Proteotoxicity
2.3. Molecular Insights from Affected Tissues
3. Molecular Features of Amyloid LCs Associated with Organ Damage
3.1. Proteolysis and Other PTMs in AL Pathogenesis
3.2. Molecular Bases of Organ Tropism
3.3. Relation between the Amyloidogenicity and Proteotoxicity of LCs
4. Basic Research Provides Hints towards Treatment Targets against Organ Damage
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Palladini, G.; Milani, P.; Merlini, G. Management of AL amyloidosis in 2020. Blood 2020, 136, 2620–2627. [Google Scholar] [CrossRef]
- Palladini, G.; Merlini, G. How I Treat AL Amyloidosis. Blood 2021. [Google Scholar] [CrossRef]
- Mead, G.P.; Carr-Smith, H.D.; Bradwell, A.R. Free light chains. Ann. Clin. Biochem. 2008, 45, 444. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Prim. 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Milani, P.; Binder, M.; Basset, M.; Tandon, N.; Foli, A.; Dispenzieri, A.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; et al. A validated composite organ and hematologic response model for early assessment of treatment outcomes in light chain amyloidosis. Blood Cancer J. 2020, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Fazio, F.; Basset, M.; Berno, T.; LaRocca, A.; Foli, A.; Riva, M.; Benigna, F.; Oliva, S.; Nuvolone, M.; et al. High rate of profound clonal and renal responses with daratumumab treatment in heavily pre-treated patients with light chain (AL) amyloidosis and high bone marrow plasma cell infiltrate. Am. J. Hematol. 2020, 95, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Basset, M.; Curci, P.; Foli, A.; Rizzi, R.; Nuvolone, M.; Guido, R.; Gesualdo, L.; Specchia, G.; Merlini, G.; et al. Daratumumab in light chain deposition disease: Rapid and profound hematologic response preserves kidney function. Blood Adv. 2020, 4, 1321–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basset, M.; Nuvolone, M.; Palladini, G.; Merlini, G. Novel challenges in the management of immunoglobulin light chain amyloidosis: From the bench to the bedside. Expert Rev. Hematol. 2020, 13, 1003–1015. [Google Scholar] [CrossRef]
- Palladini, G.; Paiva, B.; Wechalekar, A.; Massa, M.; Milani, P.; Lasa, M.; Ravichandran, S.; Krsnik, I.; Basset, M.; Burgos, L.; et al. Minimal residual disease negativity by next-generation flow cytometry is associated with improved organ response in AL amyloidosis. Blood Cancer J. 2021, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Lavatelli, F.; Russo, P.; Perlini, S.; Perfetti, V.; Bosoni, T.; Obici, L.; Bradwell, A.; D’Eril, G.M.; Fogari, R.; et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood 2006, 107, 3854–3858. [Google Scholar] [CrossRef]
- Sarosiek, S.; Varga, C.; Jacob, A.; Fulciniti, M.T.; Munshi, N.; Sanchorawala, V. Detection of minimal residual disease by next generation sequencing in AL amyloidosis. Blood Cancer J. 2021, 11, 117. [Google Scholar] [CrossRef]
- Li, X.; Huang, B.; Liu, J.; Chen, M.; Gu, J.; Li, J. Clinical value of minimal residual disease assessed by multiparameter flow cytometry in amyloid light chain amyloidosis. J. Cancer Res. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Vaxman, I.; Gertz, M.A. Measurable residual disease in multiple myeloma and light chain amyloidosis: More than meets the eye. Leuk. Lymphoma 2021, 62, 1544–1553. [Google Scholar] [CrossRef]
- Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Lacy, M.Q.; Leung, N.; Buadi, F.K.; Dingli, D.; Hayman, S.R.; Go, R.S.; Kapoor, P.; et al. Characterization and prognostic implication of delayed complete response in AL amyloidosis. Eur. J. Haematol. 2021, 106, 354–361. [Google Scholar] [CrossRef]
- Sidana, S.; Muchtar, E.; Sidiqi, M.H.; Jevremovic, D.; Dispenzieri, A.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Hayman, S.R.; Kourelis, T.; et al. Impact of minimal residual negativity using next generation flow cytometry on outcomes in light chain amyloidosis. Am. J. Hematol. 2020, 95, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Ćorović, A.; Scully, P.; Moon, J.C. Myocardial Amyloidosis: The Exemplar Interstitial Disease. JACC Cardiovasc. Imaging 2019, 12, 2345–2356. [Google Scholar] [CrossRef]
- Kotecha, T.; Martinez-Naharro, A.; Treibel, T.; Francis, R.; Nordin, S.; Abdel-Gadir, A.; Knight, D.S.; Zumbo, G.; Rosmini, S.; Maestrini, V.; et al. Myocardial Edema and Prognosis in Amyloidosis. J. Am. Coll. Cardiol. 2018, 71, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Bellotti, V.; Corazza, A.; Foglia, B.; Novo, E.; Simons, J.P.; Mangione, P.; Verona, G.; Canetti, D.; Nocerino, P.; Obici, L.; et al. Amyloid damage to islet β-cells in type 2 diabetes: Hypoxia or pseudo-hypoxia? bioRxiv 2019, 810747. [Google Scholar] [CrossRef]
- Schleeger, M.; Vandenakker, C.C.; Deckert-Gaudig, T.; Deckert, V.; Velikov, K.P.; Koenderink, G.; Bonn, M. Amyloids: From molecular structure to mechanical properties. Polymer 2013, 54, 2473–2488. [Google Scholar] [CrossRef] [Green Version]
- Marin-Argany, M.; Lin, Y.; Misra, P.; Williams, A.; Wall, J.S.; Howell, K.G.; Elsbernd, L.R.; McClure, M.; Ramirez-Alvarado, M. Cell Damage in Light Chain Amyloidosis: Fibril Internalization, Toxicity and Cell-Mediated Seeding. J. Biol. Chem. 2016, 291, 19813–19825. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.B.; Williams, A.; Wooliver, C.; Heidel, R.E.; Adams, S.; Dunlap, J.; Ramirez-Alvarado, M.; Blancas-Mejia, L.; Kennel, S.J.; Wall, J.S. Recruitment of human light chain proteins by synthetic fibrils is dependent on disease state and may be used to predict amyloidogenic propensity. Amyloid 2017, 24, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. The Ultrastructure of Tissue Damage by Amyloid Fibrils. Molecules 2021, 26, 4611. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.L.; Maar, K.; Redhage, K.R.; Misra, P.; Blancas-Mejia, L.M.; Dick, C.J.; Wall, J.; Williams, A.; Dietz, A.B.; van Wijnen, A.J.; et al. Light chain amyloidosis induced inflammatory changes in cardiomyocytes and adipose-derived mesenchymal stromal cells. Leukemia 2020, 34, 1383–1393. [Google Scholar] [CrossRef]
- Orini, M.; Graham, A.J.; Martinez-Naharro, A.; Andrews, C.M.; De Marvao, A.; Statton, B.; Cook, S.A.; O’Regan, D.P.; Hawkins, P.N.; Rudy, Y.; et al. Noninvasive Mapping of the Electrophysiological Substrate in Cardiac Amyloidosis and Its Relationship to Structural Abnormalities. J. Am. Heart Assoc. 2019, 8, e012097. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Milani, P.; Merlini, G. In search of the most effective therapy for light chain amyloidosis. Amyloid 2021, 1–2. [Google Scholar] [CrossRef]
- Sapp, V.; Jain, M.; Liao, R. Viewing Extrinsic Proteotoxic Stress Through the Lens of Amyloid Cardiomyopathy. Physiology (Bethesda) 2016, 31, 294–299. [Google Scholar] [CrossRef]
- Liao, R.; Jain, M.; Teller, P.; Connors, L.; Ngoy, S.; Skinner, M.; Falk, R.H.; Apstein, C.S. Infusion of Light Chains From Patients With Cardiac Amyloidosis Causes Diastolic Dysfunction in Isolated Mouse Hearts. Circulation 2001, 104, 1594–1597. [Google Scholar] [CrossRef]
- Lavatelli, F.; Imperiini, E.; Orrù, S.; Rognoni, P.; Sarnataro, D.; Palladini, G.; Malpasso, G.; Soriano, M.E.; Di Fonzo, A.; Valentini, V.; et al. Novel mitochondrial protein interactors of immunoglobulin light chains causing heart amyloidosis. FASEB J. 2015, 29, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Mishra, S.; Qiu, Y.; Shi, J.; Trudeau, K.; Las, G.; Liesa, M.; Shirihai, O.S.; Connors, L.H.; Seldin, D.C.; et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol. Med. 2014, 6, 1493–1507. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; del Monte, F.; Ward, J.; Connors, L.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38 MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomede, L.; Rognoni, P.; Lavatelli, F.; Romeo, M.; DEL Favero, E.; Cantù, L.; Ghibaudi, E.; Di Fonzo, A.; Corbelli, A.; Fiordaliso, F.; et al. A Caenorhabditis elegans–based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood 2014, 123, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Merlini, G.; Palladini, G. Light chain amyloidosis: The heart of the problem. Haematologica 2013, 98, 1492–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, L.; Orfanelli, U.; Resnati, M.; Raimondi, A.; Orsi, A.; Milan, E.; Palladini, G.; Milani, P.; Cerruti, F.; Cascio, P.; et al. The amyloidogenic light chain is a stressor that sensitizes plasma cells to proteasome inhibitor toxicity. Blood 2017, 129, 2132–2142. [Google Scholar] [CrossRef]
- Cooley, C.B.; Ryno, L.M.; Plate, L.; Morgan, G.J.; Hulleman, J.D.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc. Natl. Acad. Sci. USA 2014, 111, 13046–13051. [Google Scholar] [CrossRef] [Green Version]
- Monis, G.F.; Schultz, C.; Ren, R.; Eberhard, J.; Costello, C.; Connors, L.; Skinner, M.; Trinkaus-Randall, V. Role of Endocytic Inhibitory Drugs on Internalization of Amyloidogenic Light Chains by Cardiac Fibroblasts. Am. J. Pathol. 2006, 169, 1939–1952. [Google Scholar] [CrossRef] [Green Version]
- Trinkaus-Randall, V.; Walsh, M.T.; Steeves, S.; Monis, G.; Connors, L.H.; Skinner, M. Cellular Response of Cardiac Fibroblasts to Amyloidogenic Light Chains. Am. J. Pathol. 2005, 166, 197–208. [Google Scholar] [CrossRef]
- Imperlini, E.; Gnecchi, M.; Rognoni, P.; Sabidó, E.; Ciuffreda, M.C.; Palladini, G.; Espadas, G.; Mancuso, F.; Bozzola, M.; Malpasso, G.; et al. Proteotoxicity in cardiac amyloidosis: Amyloidogenic light chains affect the levels of intracellular proteins in human heart cells. Sci. Rep. 2017, 7, 15661. [Google Scholar] [CrossRef]
- Guan, J.; Mishra, S.; Shi, J.; Plovie, E.; Qiu, Y.; Cao, X.; Gianni, D.; Jiang, B.; Del Monte, F.; Connors, L.H.; et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res. Cardiol. 2013, 108, 378. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.A.; Jain, M.; Pimentel, D.R.; Wang, B.; Connors, L.H.; Skinner, M.; Apstein, C.S.; Liao, R. Human Amyloidogenic Light Chains Directly Impair Cardiomyocyte Function Through an Increase in Cellular Oxidant Stress. Circ. Res. 2004, 94, 1008–1010. [Google Scholar] [CrossRef] [Green Version]
- Diomede, L.; Romeo, M.; Rognoni, P.; Beeg, M.; Foray, C.; Ghibaudi, E.; Palladini, G.; Cherny, R.A.; Verga, L.; Capello, G.L.; et al. Cardiac Light Chain Amyloidosis: The Role of Metal Ions in Oxidative Stress and Mitochondrial Damage. Antioxid. Redox Signal. 2017, 27, 567–582. [Google Scholar] [CrossRef]
- Mishra, S.; Guan, J.; Plovie, E.; Seldin, D.C.; Connors, L.; Merlini, G.; Falk, R.H.; Macrae, C.A.; Liao, R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am. J. Physiol. Circ. Physiol. 2013, 305, H95–H103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Joshi, S.; Ward, J.E.; Buys, E.P.; Mishra, D.; Morgado, I.; Fisch, S.; Lavatelli, F.; Merlini, G.; Dorbala, S.; et al. Zebrafish model of amyloid light chain cardiotoxicity: Regeneration versus degeneration. Am. J. Physiol. Circ. Physiol. 2019, 316, H1158–H1166. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.E.; Ren, R.; Toraldo, G.; Soohoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef] [Green Version]
- Nuvolone, M.; Sorce, S.; Pelczar, P.; Rushing, E.; Lavatelli, F.; Rognoni, P.; Valentini, V.; Palladini, G.; Merlini, G.; Aguzzi, A. Regulated expression of amyloidogenic immunoglobulin light chains in mice. Amyloid 2017, 24, 52–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhou, P.; Kugelmass, A.; Toskic, D.; Warner, M.; Lee, L.; Fogaren, T.; Godara, A.; Wang, M.; Li, Y.; et al. A novel xenograft mouse model for testing approaches targeting human kappa light-chain diseases. Gene Ther. 2019, 26, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Sirac, C.; Herrera, G.A.; Sanders, P.W.; Batuman, V.; Bender, S.; Ayala, M.V.; Javaugue, V.; Teng, J.; Turbat-Herrera, E.A.; Cogné, M.; et al. Animal models of monoclonal immunoglobulin-related renal diseases. Nat. Rev. Nephrol. 2018, 14, 246–264. [Google Scholar] [CrossRef]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. An animal model of glomerular light-chain-associated amyloidogenesis depicts the crucial role of lysosomes. Kidney Int. 2014, 86, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Herrera, G.A.; Turbat-Herrera, E.A.; Teng, J. Animal model of renal AL-amyloidogenesis recapitulates in vitro findings. Amyloid 2011, 18 (Suppl. 1), 34–37. [Google Scholar] [CrossRef]
- Merlini, G.; Lousada, I.; Ando, Y.; Dispenzieri, A.; Gertz, M.A.; Grogan, M.; Maurer, M.S.; Sanchorawala, V.; Wechalekar, A.; Palladini, G.; et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia 2016, 30, 1979–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maritan, M.; Ambrosetti, A.; Oberti, L.; Barbiroli, A.; Diomede, L.; Romeo, M.; Lavatelli, F.; Sormanni, P.; Palladini, G.; Bolognesi, M.; et al. Modulating the cardiotoxic behaviour of immunoglobulin light chain dimers through point mutations. Amyloid 2019, 26, 105–106. [Google Scholar] [CrossRef]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. Extrusion of Amyloid Fibrils to the Extracellular Space in Experimental Mesangial AL-Amyloidosis: Transmission and Scanning Electron Microscopy Studies and Correlation with Renal Biopsy Observations. Ultrastruct. Pathol. 2013, 38, 104–115. [Google Scholar] [CrossRef]
- Herrera, G.A.; del Pozo-Yauner, L.; Teng, J.; Zeng, C.; Shen, X.; Moriyama, T.; Alcantara, V.R.; Liu, B.; Turbat-Herrera, E.A. Glomerulopathic Light Chain-Mesangial Cell Interactions: Sortilin-Related Receptor (SORL1) and Signaling. Kidney Int. Rep. 2021, 6, 1379–1396. [Google Scholar] [CrossRef]
- Herrera, G.A.; Teng, J.; Turbat-Herrera, E.A.; Zeng, C.; del Pozo-Yauner, L. Understanding Mesangial Pathobiology in AL-Amyloidosis and Monoclonal Ig Light Chain Deposition Disease. Kidney Int. Rep. 2020, 5, 1870–1893. [Google Scholar] [CrossRef] [PubMed]
- Truran, S.; Weissig, V.; Ramirez-Alvarado, M.; Franco, D.A.; Burciu, C.; Georges, J.; Murarka, S.; Okoth, W.A.; Schwab, S.; Hari, P.; et al. Nanoliposomes protect against AL amyloid light chain protein-induced endothelial injury. J. Liposome Res. 2013, 24, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, D.A.; Truran, S.; Burciu, C.; Gutterman, D.D.; Maltagliati, A.; Weissig, V.; Hari, P.; Migrino, R.Q. Protective role of clusterin in preserving endothelial function in AL amyloidosis. Atherosclerosis 2012, 225, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migrino, R.Q.; Truran, S.; Gutterman, D.D.; Franco, D.A.; Bright, M.; Schlundt, B.; Timmons, M.; Motta, A.; Phillips, S.A.; Hari, P. Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins. Am. J. Physiol. Circ. Physiol. 2011, 301, H2305–H2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migrino, R.Q.; Hari, P.; Gutterman, D.D.; Bright, M.; Truran, S.; Schlundt, B.; Phillips, S.A. Systemic and microvascular oxidative stress induced by light chain amyloidosis. Int. J. Cardiol. 2010, 145, 67–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, F.; Lavatelli, F.; Merlini, G.; Mauri, P. Clinical proteomics for diagnosis and typing of systemic amyloidoses. Proteom. Clin. Appl. 2013, 7, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Palladini, G.; Merlini, G.; Mauri, P. Shotgun Protein Profile of Human Adipose Tissue and Its Changes in Relation to Systemic Amyloidoses. J. Proteome Res. 2013, 12, 5642–5655. [Google Scholar] [CrossRef]
- Lavatelli, F.; Perlman, D.H.; Spencer, B.; Prokaeva, T.; McComb, M.E.; Théberge, R.; Connors, L.; Bellotti, V.; Seldin, D.C.; Merlini, G.; et al. Amyloidogenic and Associated Proteins in Systemic Amyloidosis Proteome of Adipose Tissue. Mol. Cell. Proteom. 2008, 7, 1570–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Rossi, R.; Palladini, G.; Obici, L.; Verga, L.; Mauri, P.; Merlini, G. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012, 119, 1844–1847. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, T.V.; Dasari, S.S.; Dispenzieri, A.; Maleszewski, J.J.; Redfield, M.M.; Fayyaz, A.U.; Grogan, M.; Ramirez-Alvarado, M.; Ezzeddine, O.F.A.; McPhail, E.D. A Proteomic Atlas of Cardiac Amyloid Plaques. JACC CardioOncol. 2020, 2, 632–643. [Google Scholar] [CrossRef]
- Tanaka, K.; Essick, E.E.; Doros, G.; Tanriverdi, K.; Connors, L.H.; Seldin, D.C.; Sam, F. Circulating Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Cardiac Amyloidosis. J. Am. Heart Assoc. 2013, 2, e005868. [Google Scholar] [CrossRef] [Green Version]
- Bohne, S.; Sletten, K.; Menard, R.; Bühling, F.; Vöckler, S.; Wrenger, E.; Roessner, A.; Röcken, C. Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J. Pathol. 2004, 203, 528–537. [Google Scholar] [CrossRef]
- Röcken, C.; Stix, B.; Brömme, D.; Ansorge, S.; Roessner, A.; Bühling, F. A Putative Role for Cathepsin K in Degradation of AA and AL Amyloidosis. Am. J. Pathol. 2001, 158, 1029–1038. [Google Scholar] [CrossRef]
- Richey, T.; Foster, J.S.; Williams, A.D.; Williams, A.; Stroh, A.; Macy, S.; Wooliver, C.; Heidel, R.E.; Varanasi, S.K.; Ergen, E.N.; et al. Macrophage-Mediated Phagocytosis and Dissolution of Amyloid-Like Fibrils in Mice, Monitored by Optical Imaging. Am. J. Pathol. 2019, 189, 989–998. [Google Scholar] [CrossRef]
- Ami, D.; Lavatelli, F.; Rognoni, P.; Palladini, G.; Raimondi, S.; Giorgetti, S.; Monti, L.; Doglia, S.M.; Natalello, A.; Merlini, G. In situ characterization of protein aggregates in human tissues affected by light chain amyloidosis: A FTIR microspectroscopy study. Sci. Rep. 2016, 6, 29096. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, G.; Ricagno, S.; Caminito, S.; Rognoni, P.; Milani, P.; Nuvolone, M.; Basset, M.; Foli, A.; Russo, R.; Merlini, G.; et al. Protease-sensitive regions in amyloid light chains: What a common pattern of fragmentation across organs suggests about aggregation. FEBS J. 2021. [Google Scholar] [CrossRef]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: Insights into the timing of proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2018, 33, 254–257. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Jiang, Y.; Budnik, M.; Théberge, R.; Prokaeva, T.; Bodi, K.L.; Seldin, D.C.; Costello, C.E.; Skinner, M. Heterogeneity in primary structure, post-translational modifications, and germline gene usage of nine full-length amyloidogenic kappa1 immunoglobulin light chains. Biochemistry 2007, 46, 14259–14271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Misra, P.; Blancas-Mejia, L.M.; Ramirez-Alvarado, M. Mechanistic Insights into the Early Events in the Aggregation of Immunoglobulin Light Chains. Biochemistry 2019, 58, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Martin, E.B.; Williams, A.; Wall, J.S.; Ramirez-Alvarado, M. Kinetic stability and sequence/structure studies of urine-derived Bence-Jones proteins from multiple myeloma and light chain amyloidosis patients. Biophys. Chem. 2017, 230, 89–98. [Google Scholar] [CrossRef]
- González-Andrade, M.; Becerril-Luján, B.; Sánchez-López, R.; Ceceña-Álvarez, H.; Pérez-Carreón, J.I.; Ortiz, E.; Fernandez-Velasco, D.A.; Del Pozo-Yauner, L. Mutational and genetic determinants of λ6 light chain amyloidogenesis. FEBS J. 2013, 280, 6173–6183. [Google Scholar] [CrossRef]
- Ruiz-Zamora, R.A.; Guillaumé, S.; Al-Hilaly, Y.K.; Al-Garawi, A.P.D.Z.; Rodríguez-Alvarez, F.J.; Zavala-Padilla, G.; Pérez-Carreón, J.I.; Rodríguez-Ambriz, S.L.; Herrera, G.A.; Becerril-Luján, B.; et al. The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation. Sci. Rep. 2019, 9, 3123. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Santoyo, A.; del Pozo Yauner, L.; Fuentes-Silva, D.; Ortiz, E.; Rudiño-Piñera, E.; Sánchez-López, R.; Horjales, E.; Becerril, B.; Rodríguez-Romero, A. A single mutation at the sheet switch region results in conformational changes favoring lambda6 light-chain fibrillogenesis. J. Mol. Biol. 2010, 396, 280–292. [Google Scholar] [CrossRef]
- del Pozo-Yauner, L.; Ortiz, E.; Sánchez, R.; Sanchez-Lopez, R.; Güereca, L.; Murphy, C.L.; Allen, A.; Wall, J.; Fernández-Velasco, D.A.; Solomon, A.; et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins Struct. Funct. Bioinform. 2008, 72, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.-T.; Cendales, M.D.P.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. eLife 2020, 9, e52300. [Google Scholar] [CrossRef]
- Garofalo, M.; Piccoli, L.; Romeo, M.; Barzago, M.M.; Ravasio, S.; Foglierini, M.; Matkovic, M.; Sgrignani, J.; De Gasparo, R.; Prunotto, M.; et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat. Commun. 2021, 12, 3532. [Google Scholar] [CrossRef]
- Rawat, P.; Prabakaran, R.; Kumar, S.; Gromiha, M.M. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci. Rep. 2021, 11, 13785. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Hora, M.; Kazman, P.; Pradhan, T.; Rührnößl, F.; Reif, B.; Buchner, J. Domain Interactions Determine the Amyloidogenicity of Antibody Light Chain Mutants. J. Mol. Biol. 2020, 432, 6187–6199. [Google Scholar] [CrossRef]
- Weber, B.; Hora, M.; Kazman, P.; Göbl, C.; Camilloni, C.; Reif, B.; Buchner, J. The Antibody Light-Chain Linker Regulates Domain Orientation and Amyloidogenicity. J. Mol. Biol. 2018, 430, 4925–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maritan, M.; Romeo, M.; Oberti, L.; Sormanni, P.; Tasaki, M.; Russo, R.; Ambrosetti, A.; Motta, P.; Rognoni, P.; Mazzini, G.; et al. Inherent Biophysical Properties Modulate the Toxicity of Soluble Amyloidogenic Light Chains. J. Mol. Biol. 2020, 432, 845–860. [Google Scholar] [CrossRef]
- Oberti, L.; Rognoni, P.; Barbiroli, A.G.; Lavatelli, F.; Russo, R.; Maritan, M.; Palladini, G.; Bolognesi, M.; Merlini, G.; Ricagno, S. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. 2017, 7, 16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrana, J.A.; Gamez, J.D.; Madden, B.J.; Theis, J.D.; Bergen, H.R.; Dogan, A. Classification of amyloidosis by laser microdissection and mass spectrometry–based proteomic analysis in clinical biopsy specimens. Blood 2009, 114, 4957–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennella, E.; Morgan, G.J.; Kelly, J.W.; Kay, L.E. Role of domain interactions in the aggregation of full-length immunoglobulin light chains. Proc. Natl. Acad. Sci. USA 2019, 116, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The Critical Role of the Constant Region in Thermal Stability and Aggregation of Amyloidogenic Immunoglobulin Light Chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; Yan, N.L.; Mortenson, D.E.; Rennella, E.; Blundon, J.M.; Gwin, R.M.; Lin, C.-Y.; Stanfield, R.L.; Brown, S.J.; Rosen, H.; et al. Stabilization of amyloidogenic immunoglobulin light chains by small molecules. Proc. Natl. Acad. Sci. USA 2019, 116, 8360–8369. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; Usher, G.; Kelly, J.W. Incomplete Refolding of Antibody Light Chains to Non-Native, Protease-Sensitive Conformations Leads to Aggregation: A Mechanism of Amyloidogenesis in Patients? Biochemistry 2017, 56, 6597–6614. [Google Scholar] [CrossRef]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radamaker, L.; Lin, Y.-H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [Green Version]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J. Aberrant immunoglobulin synthesis in light chain amyloidosis. Free light chain and light chain fragment production by human bone marrow cells in short-term tissue culture. J. Clin. Investig. 1986, 78, 798–806. [Google Scholar] [CrossRef]
- Buxbaum, J. Mechanisms of disease: Monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol. Clin. N. Am. 1992, 6, 323–346. [Google Scholar] [CrossRef]
- Lavatelli, F.; Brambilla, F.; Valentini, V.; Rognoni, P.; Casarini, S.; Di Silvestre, D.; Perfetti, V.; Palladini, G.; Sarais, G.; Mauri, P.; et al. A novel approach for the purification and proteomic analysis of pathogenic immunoglobulin free light chains from serum. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 409–419. [Google Scholar] [CrossRef]
- Olsen, K.E.; Sletten, K.; Westermark, P. Fragments of the constant region of immunoglobulin light chains are constituents of AL-amyloid proteins. Biochem. Biophys. Res. Commun. 1998, 251, 642–647. [Google Scholar] [CrossRef]
- Engvig, J.P.; Olsen, K.E.; Gislefoss, R.E.; Sletten, K.; Wahlström, O.; Westermark, P. Constant region of a kappa III immunoglobulin light chain as a major AL-amyloid protein. Scand. J. Immunol. 1998, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, Y.; Prokaeva, T.; Connors, L.H.; Costello, C.E. Oxidative post-translational modifications of an amyloidogenic immunoglobulin light chain protein. Int. J. Mass Spectrom. 2017, 416, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, V.; Palladini, G.; Casarini, S.; Navazza, V.; Rognoni, P.; Obici, L.; Invernizzi, R.; Perlini, S.; Klersy, C.; Merlini, G. The repertoire of λ light chains causing predominant amyloid heart involvement and identification of a preferentially involved germline gene, IGLV1-44. Blood 2012, 119, 144–150. [Google Scholar] [CrossRef]
- Abraham, R.S.; Geyer, S.M.; Price-Troska, T.L.; Allmer, C.; Kyle, R.A.; Gertz, M.A.; Fonseca, R. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain–associated amyloidosis (AL). Blood 2003, 101, 3801–3807. [Google Scholar] [CrossRef] [PubMed]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The tropism of organ involvement in primary systemic amyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G. Barriers to Small Molecule Drug Discovery for Systemic Amyloidosis. Molecules 2021, 26, 3571. [Google Scholar] [CrossRef]
- Yan, N.L.; Santos-Martins, D.; Nair, R.; Chu, A.; Wilson, I.A.; Johnson, K.A.; Forli, S.; Morgan, G.J.; Petrassi, H.M.; Kelly, J.W. Discovery of Potent Coumarin-Based Kinetic Stabilizers of Amyloidogenic Immunoglobulin Light Chains Using Structure-Based Design. J. Med. Chem. 2021, 64, 6273–6299. [Google Scholar] [CrossRef]
- Yan, N.L.; Santos-Martins, D.; Rennella, E.; Sanchez, B.B.; Chen, J.S.; Kay, L.E.; Wilson, I.A.; Morgan, G.J.; Forli, S.; Kelly, J.W. Structural basis for the stabilization of amyloidogenic immunoglobulin light chains by hydantoins. Bioorg. Med. Chem. Lett. 2020, 30, 127356. [Google Scholar] [CrossRef]
- Franco, D.A.; Truran, S.; Weissig, V.; Guzman-Villanueva, D.; Karamanova, N.; Senapati, S.; Burciu, C.; Ramirez-Alvarado, M.; Blancas-Mejia, L.M.; Lindsay, S.; et al. Monosialoganglioside-Containing Nanoliposomes Restore Endothelial Function Impaired by AL Amyloidosis Light Chain Proteins. J. Am. Heart Assoc. 2016, 5, e003318. [Google Scholar] [CrossRef] [Green Version]
- Godara, A.; Siddiqui, N.S.; Lee, L.X.; Toskic, D.; Fogaren, T.; Varga, C.; Comenzo, R.L. Dual Monoclonal Antibody Therapy in Patients With Systemic AL Amyloidosis and Cardiac Involvement. Clin. Lymphoma Myeloma Leuk. 2020, 20, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Varga, C.; Lentzsch, S.; Comenzo, R.L. Beyond NEOD001 for systemic light-chain amyloidosis. Blood 2018, 132, 1992–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.A.; Landau, H.; Comenzo, R.L.; Seldin, D.; Weiss, B.; Zonder, J.; Merlini, G.; Schönland, S.; Walling, J.; Kinney, G.G.; et al. First-in-Human Phase I/II Study of NEOD001 in Patients With Light Chain Amyloidosis and Persistent Organ Dysfunction. J. Clin. Oncol. 2016, 34, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Rius, B.; Mesgarzadeh, J.S.; Romine, I.C.; Paxman, R.J.; Kelly, J.W.; Wiseman, R.L. Pharmacologic targeting of plasma cell endoplasmic reticulum proteostasis to reduce amyloidogenic light chain secretion. Blood Adv. 2021, 5, 1037–1049. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Migrino, R.Q.; Truran, S.; Karamanova, N.; Franco, D.A.; Burciu, C.; Senapati, S.; Nedelkov, D.; Hari, P.; Weissig, V. PEGylated-nanoliposomal clusterin for amyloidogenic light chain-induced endothelial dysfunction. J. Liposome Res. 2017, 28, 97–105. [Google Scholar] [CrossRef]
- Andrich, K.; Hegenbart, U.; Kimmich, C.; Kedia, N.; Bergen, H.R.; Schönland, S.; Wanker, E.; Bieschke, J. Aggregation of Full-length Immunoglobulin Light Chains from Systemic Light Chain Amyloidosis (AL) Patients Is Remodeled by Epigallocatechin-3-gallate. J. Biol. Chem. 2017, 292, 2328–2344. [Google Scholar] [CrossRef] [Green Version]
- Meshitsuka, S.; Shingaki, S.; Hotta, M.; Goto, M.; Kobayashi, M.; Ukawa, Y.; Sagesaka, Y.M.; Wada, Y.; Nojima, M.; Suzuki, K. Phase 2 trial of daily, oral epigallocatechin gallate in patients with light-chain amyloidosis. Int. J. Hematol. 2016, 105, 295–308. [Google Scholar] [CrossRef]
- Mereles, D.; Buss, S.J.; Hardt, S.E.; Hunstein, W.; Katus, H.A. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin. Res. Cardiol. 2010, 99, 483–490. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Whelan, C. Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017, 7, e546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Marin-Argany, M.; Dick, C.J.; Redhage, K.R.; Blancas-Mejia, L.M.; Bulur, P.; Butler, G.W.; Deeds, M.C.; Madden, B.J.; Williams, A.; et al. Mesenchymal stromal cells protect human cardiomyocytes from amyloid fibril damage. Cytotherapy 2017, 19, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.B.; Cookson, L.M.; Barton, S.V.; Liefaard, L.; Lane, T.; Hutt, D.F.; Ritter, J.M.; Fontana, M.; Moon, J.C.; Gillmore, J.D.; et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci. Transl. Med. 2018, 10, eaan3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, D.; Cookson, L.M.; Berges, A.C.; Barton, S.V.; Lane, T.; Ritter, J.M.; Fontana, M.; Moon, J.; Pinzani, M.; Gillmore, J.D.; et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N. Engl. J. Med. 2015, 373, 1106–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavatelli, F. Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis. Hemato 2022, 3, 47-62. https://doi.org/10.3390/hemato3010005
Lavatelli F. Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis. Hemato. 2022; 3(1):47-62. https://doi.org/10.3390/hemato3010005
Chicago/Turabian StyleLavatelli, Francesca. 2022. "Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis" Hemato 3, no. 1: 47-62. https://doi.org/10.3390/hemato3010005
APA StyleLavatelli, F. (2022). Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis. Hemato, 3(1), 47-62. https://doi.org/10.3390/hemato3010005