
Effects on Microstructure and Ionic Conductivity of the Co-Doping with Strontium and Samarium of Ceria with Constant Oxygen Vacancy Concentration

Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
3.1. Composition
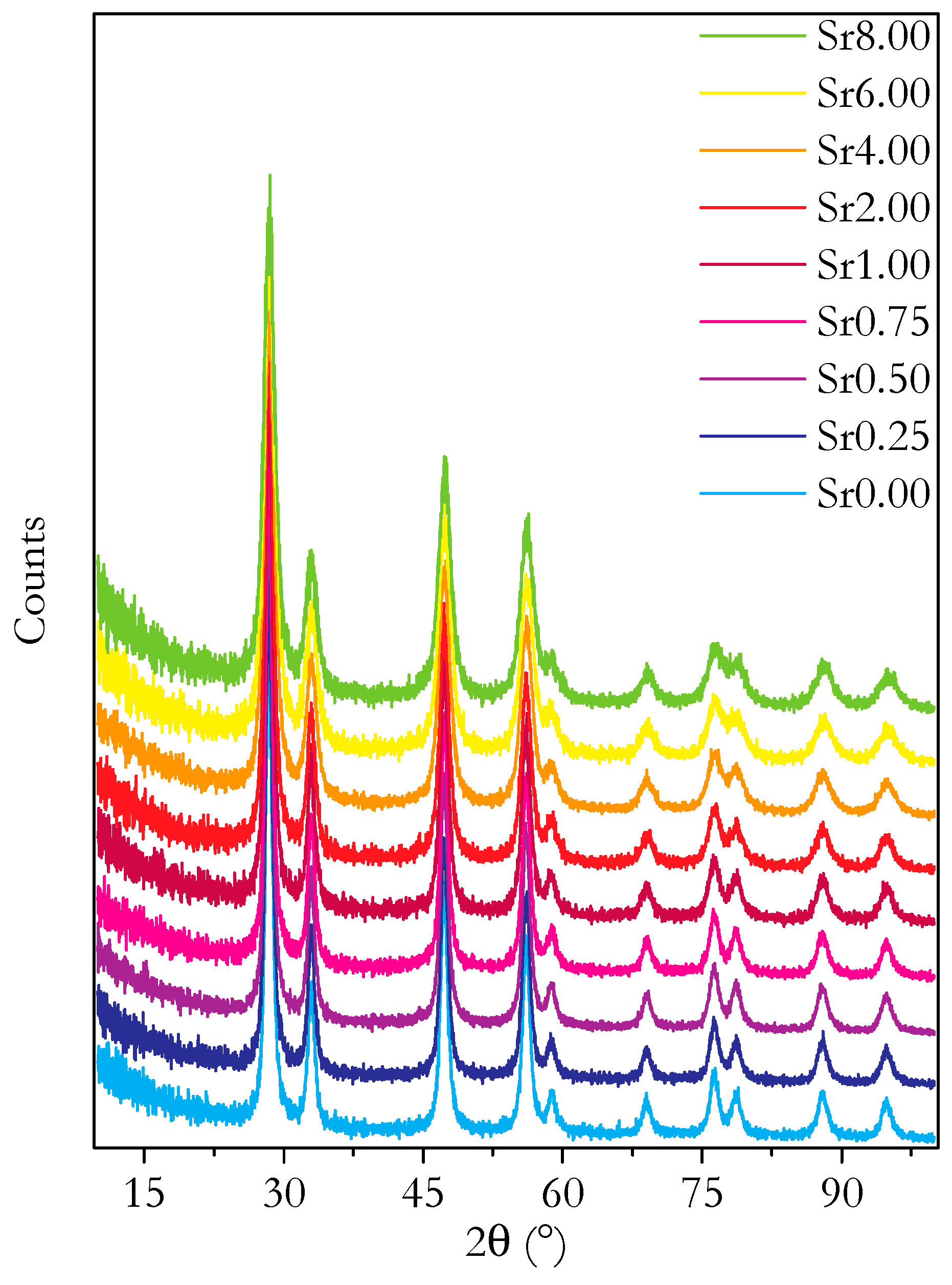
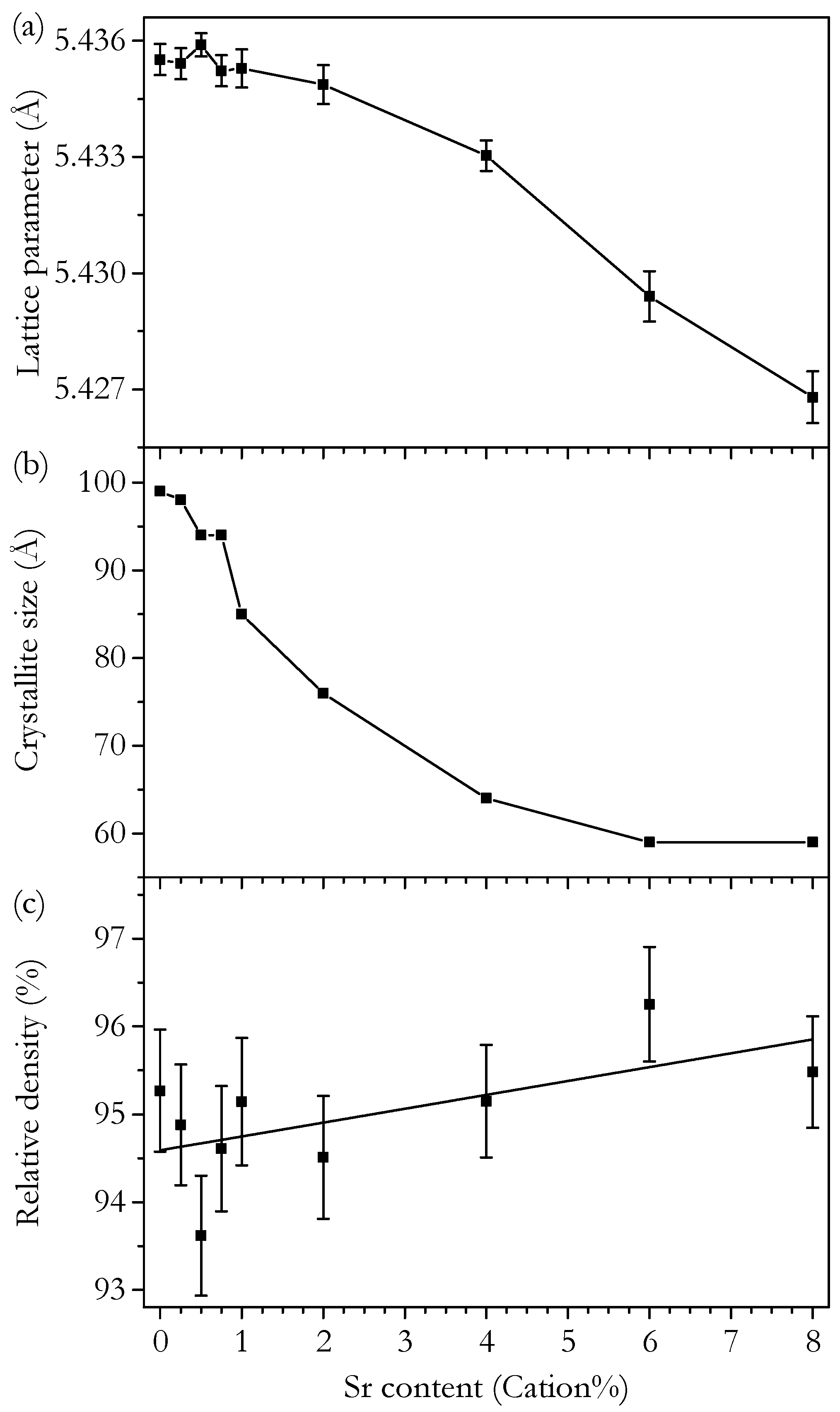
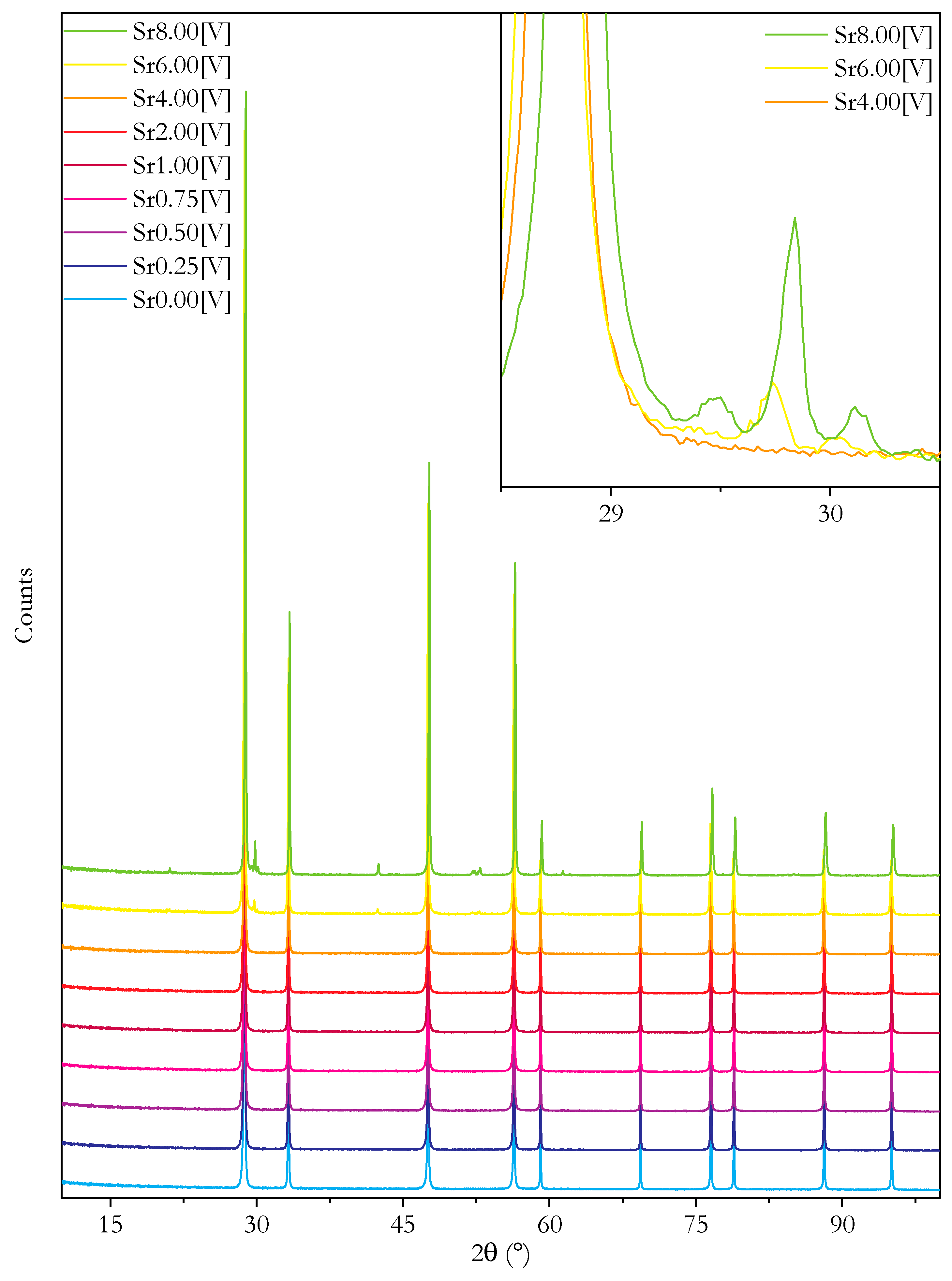
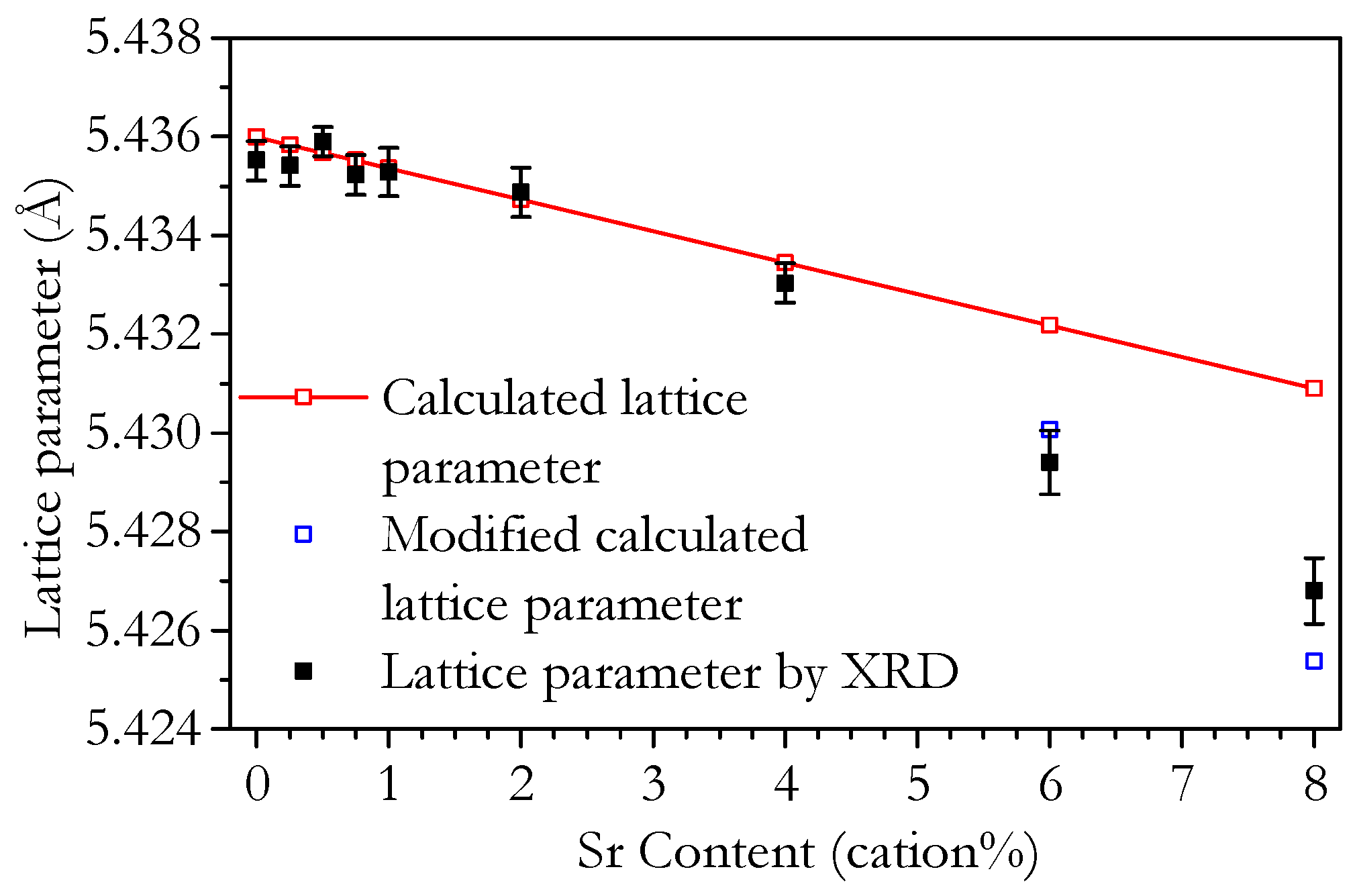
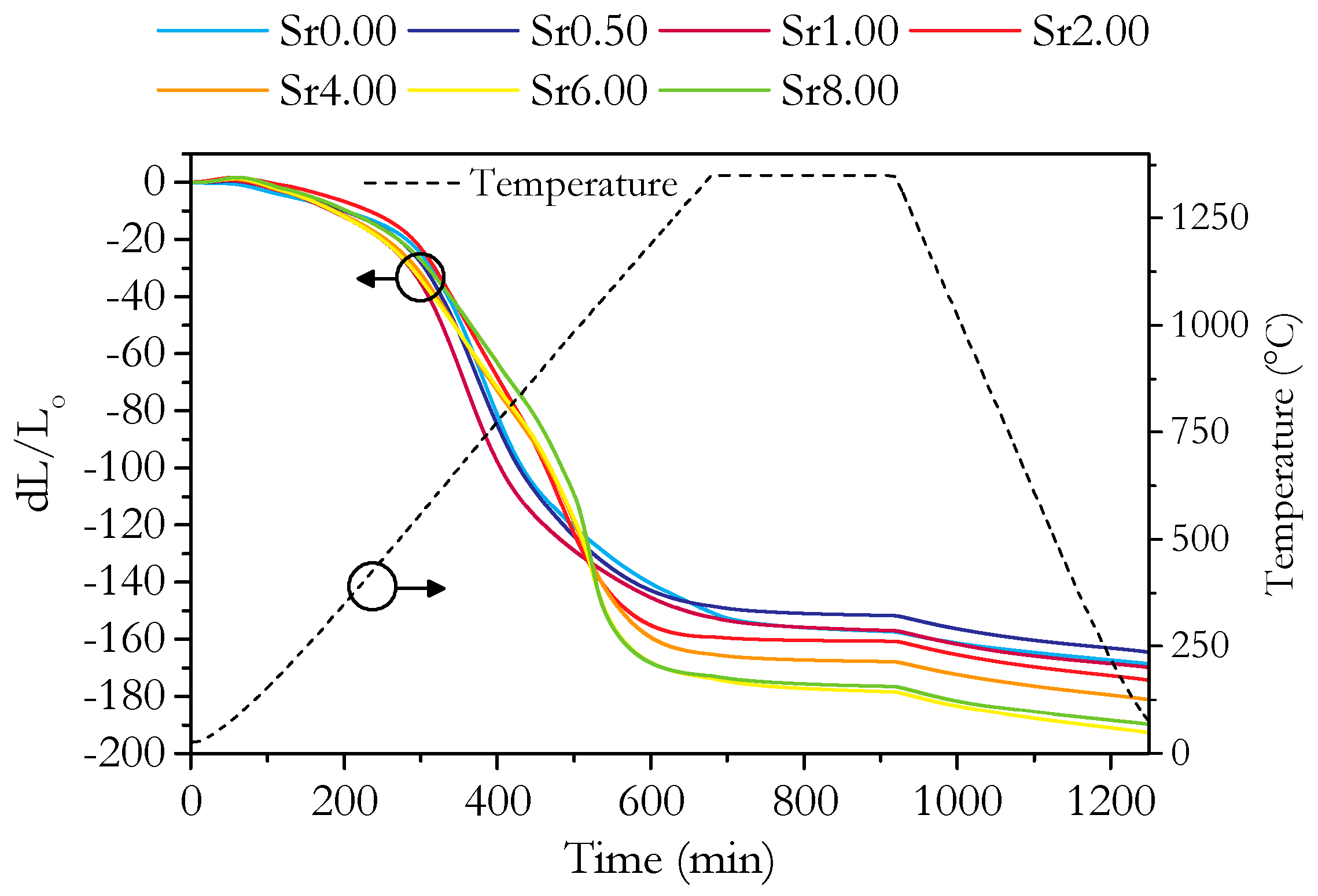
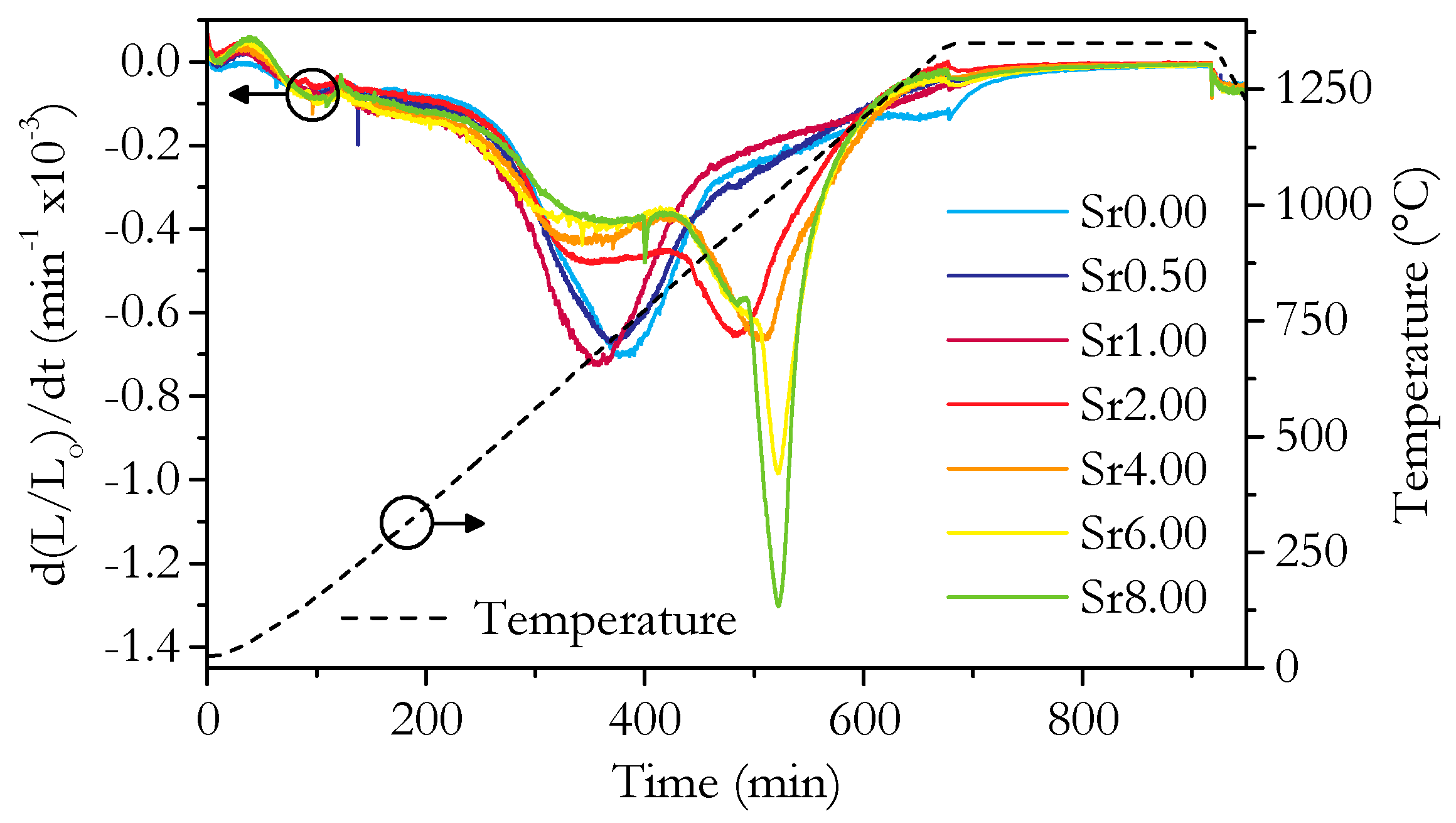
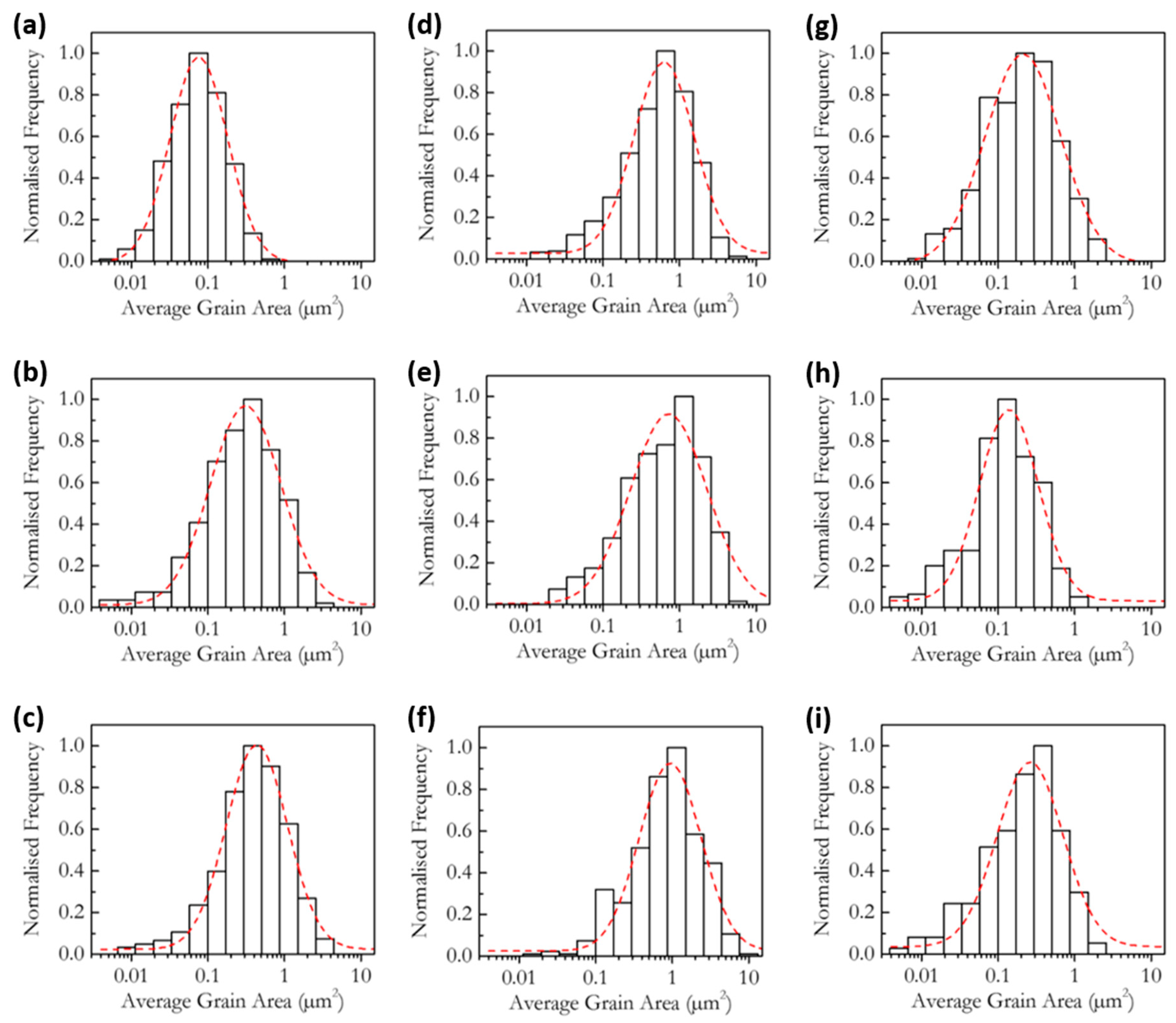
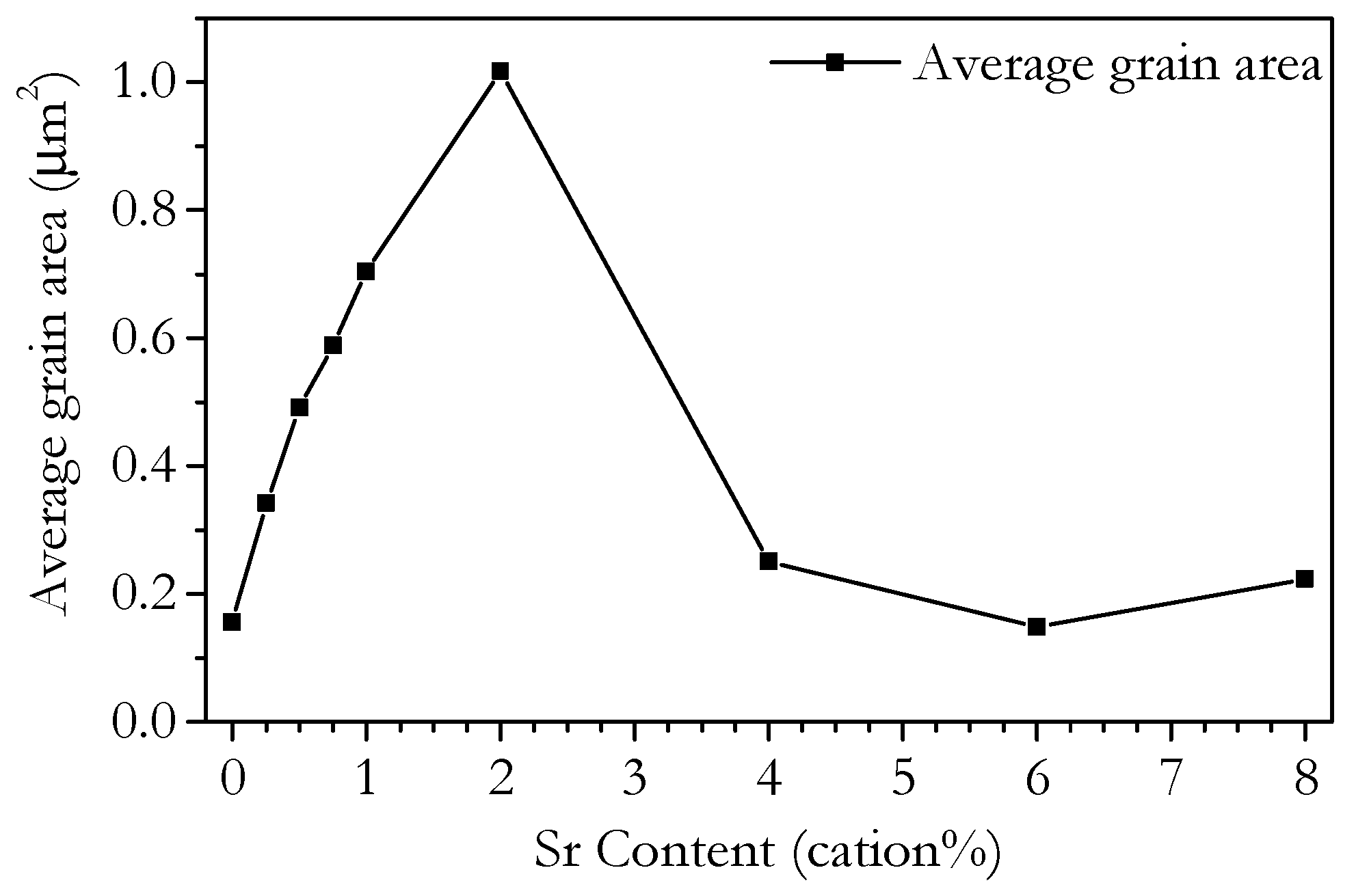
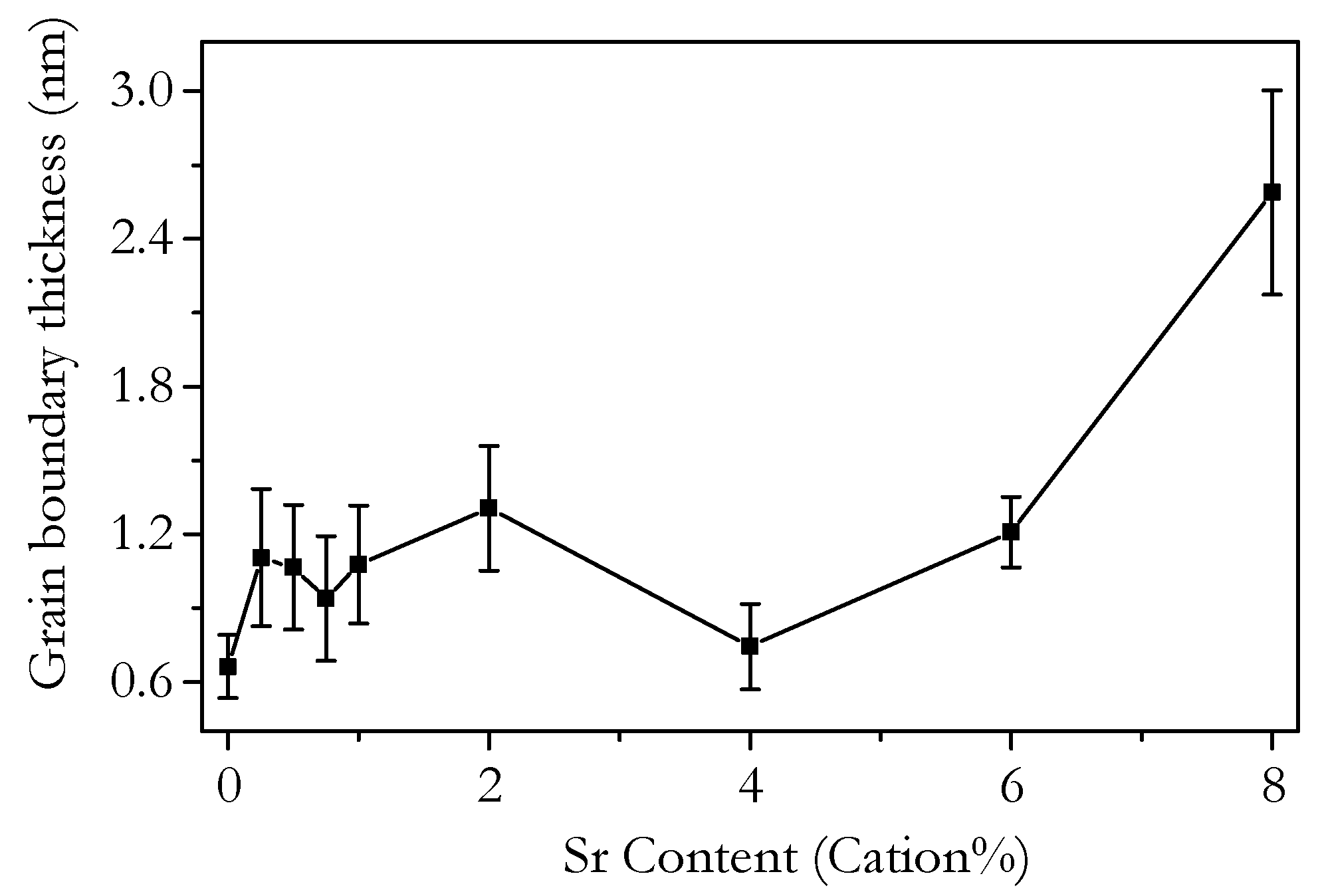
3.2. Microstructure and Phase Analysis
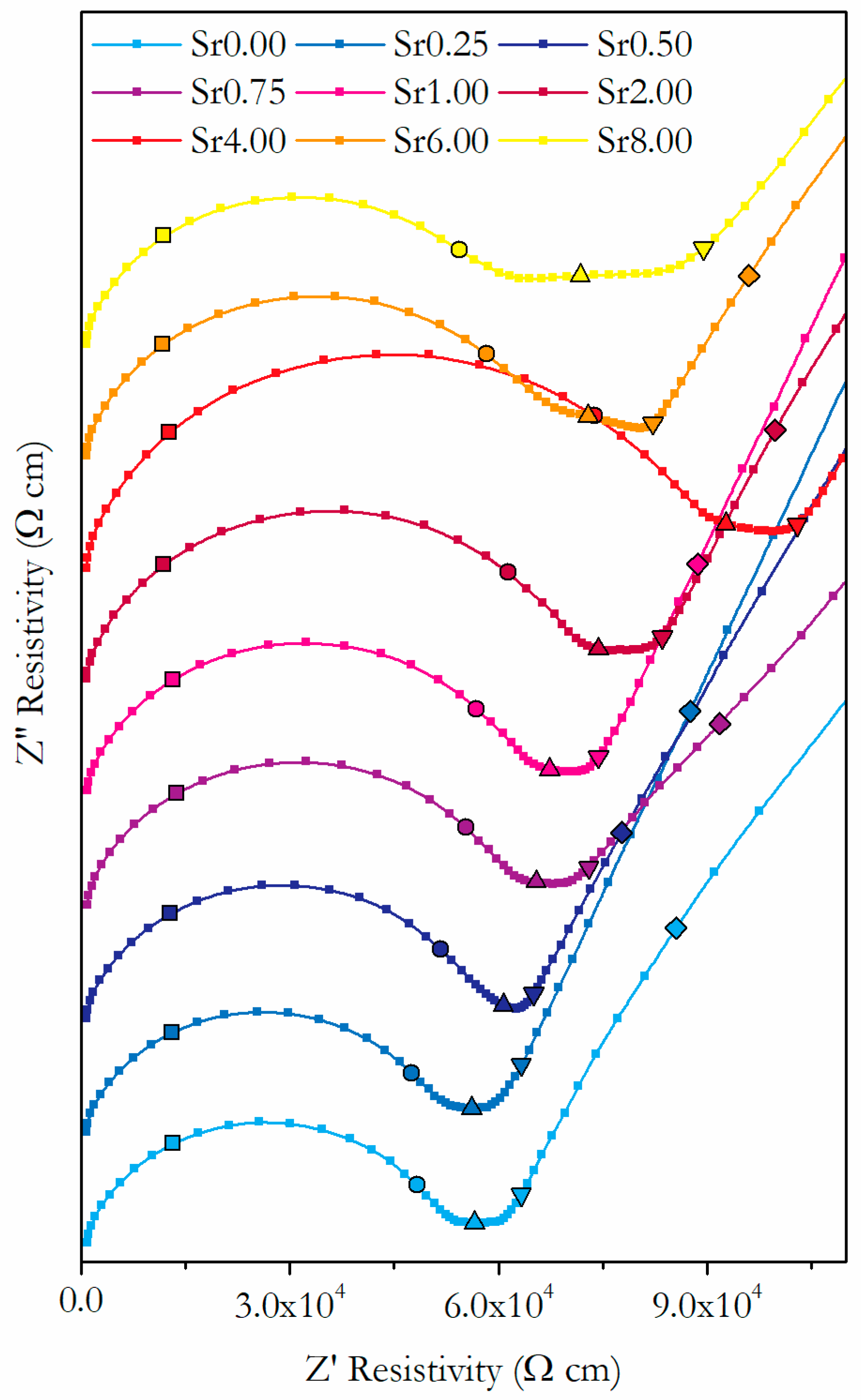
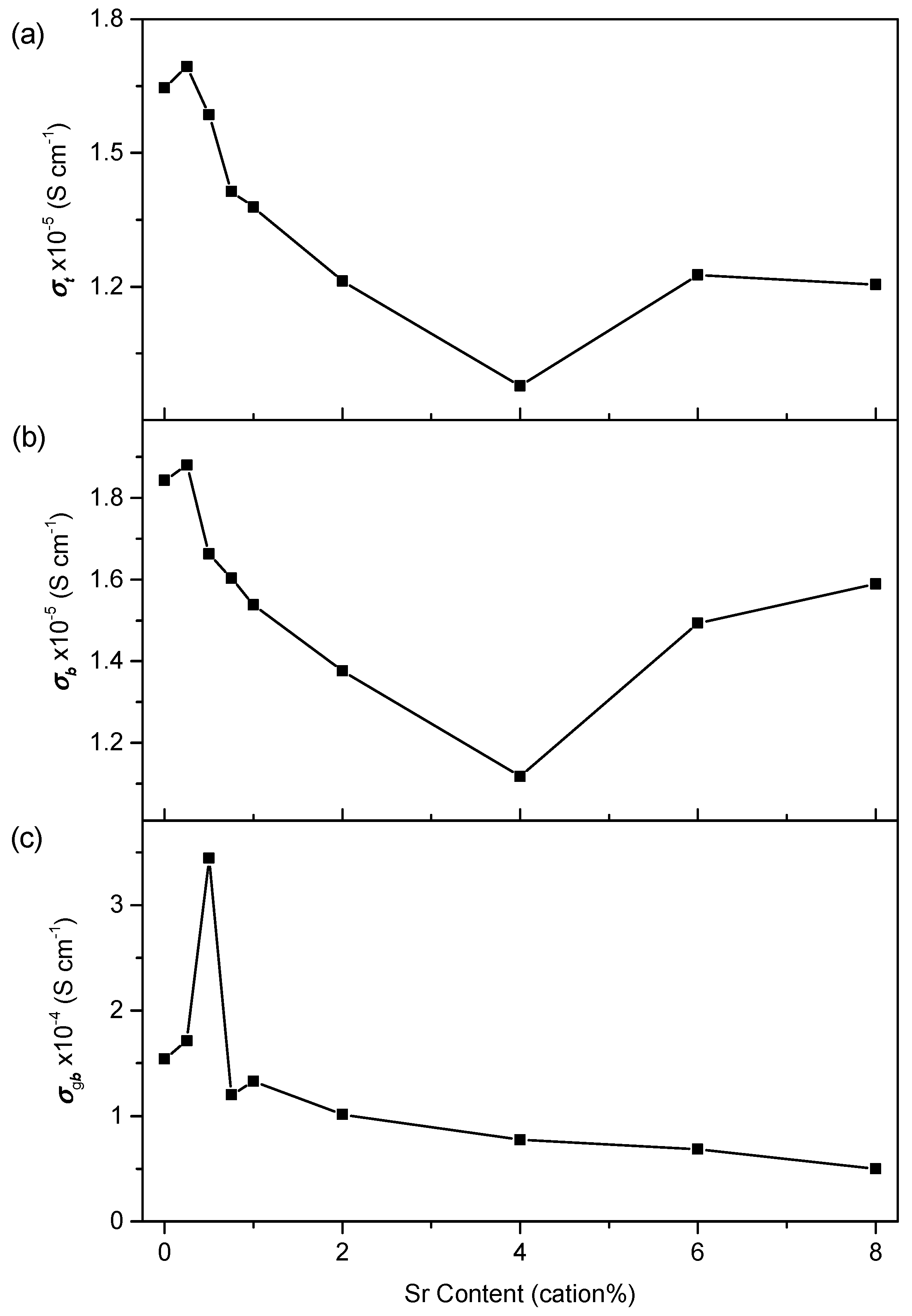
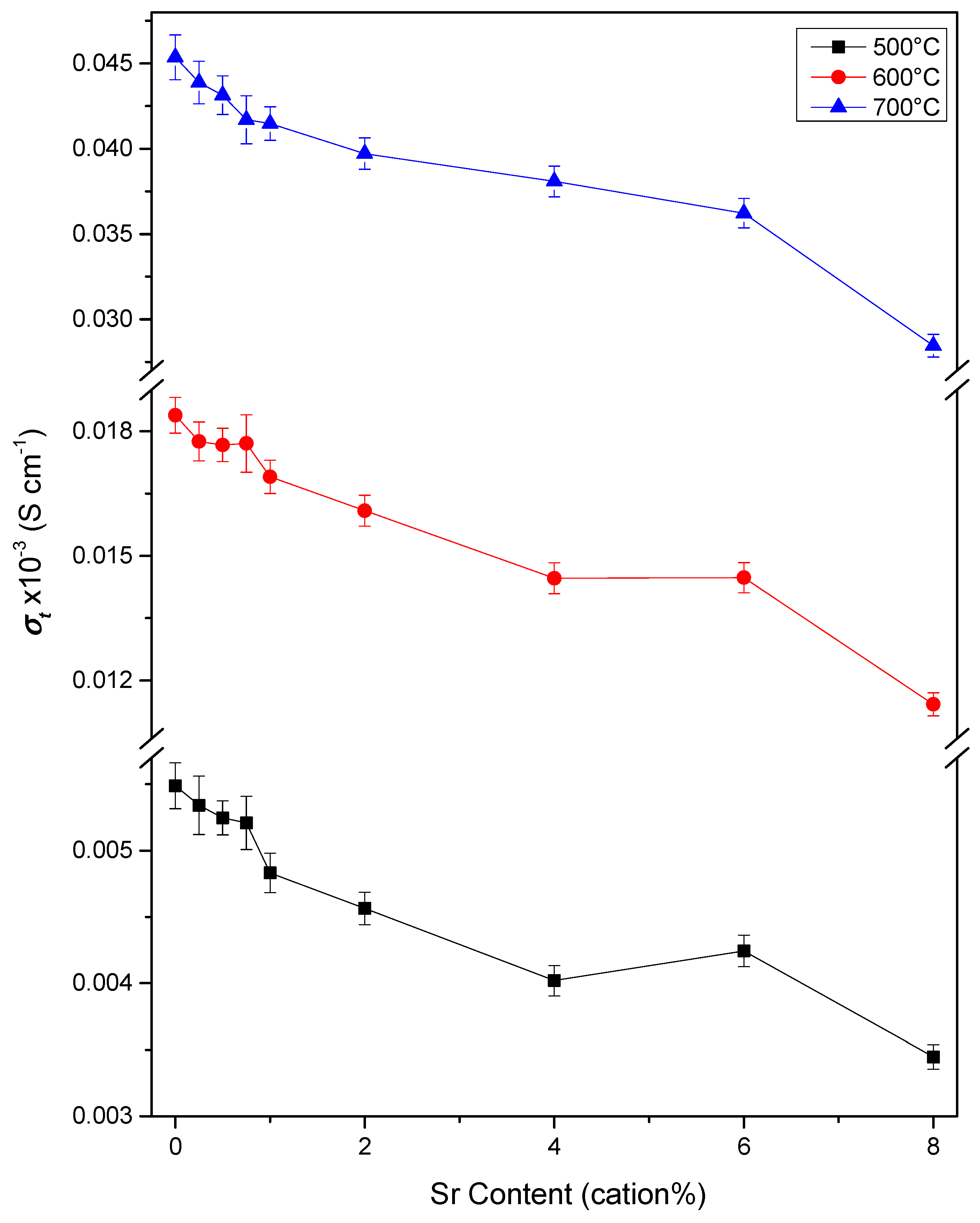
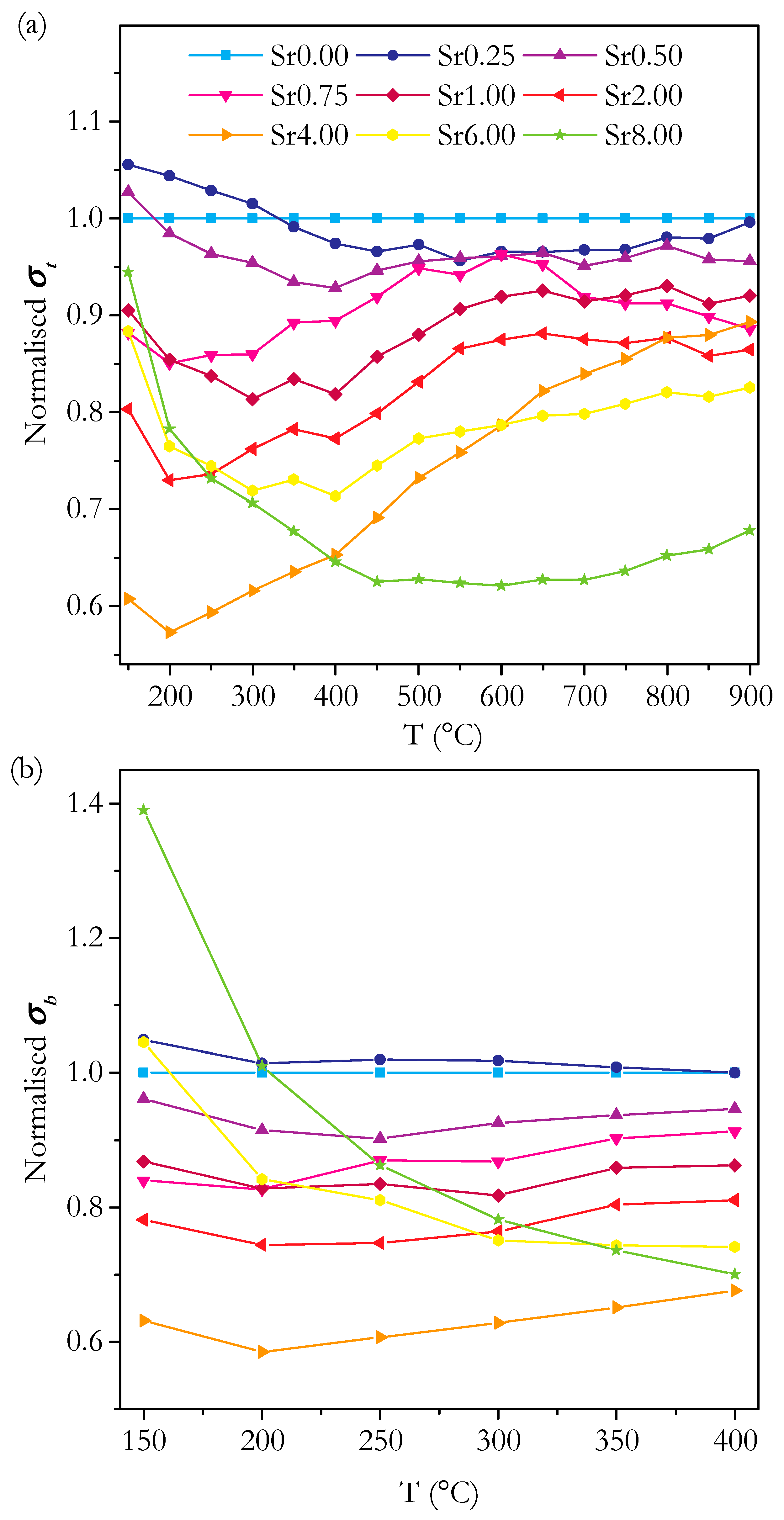
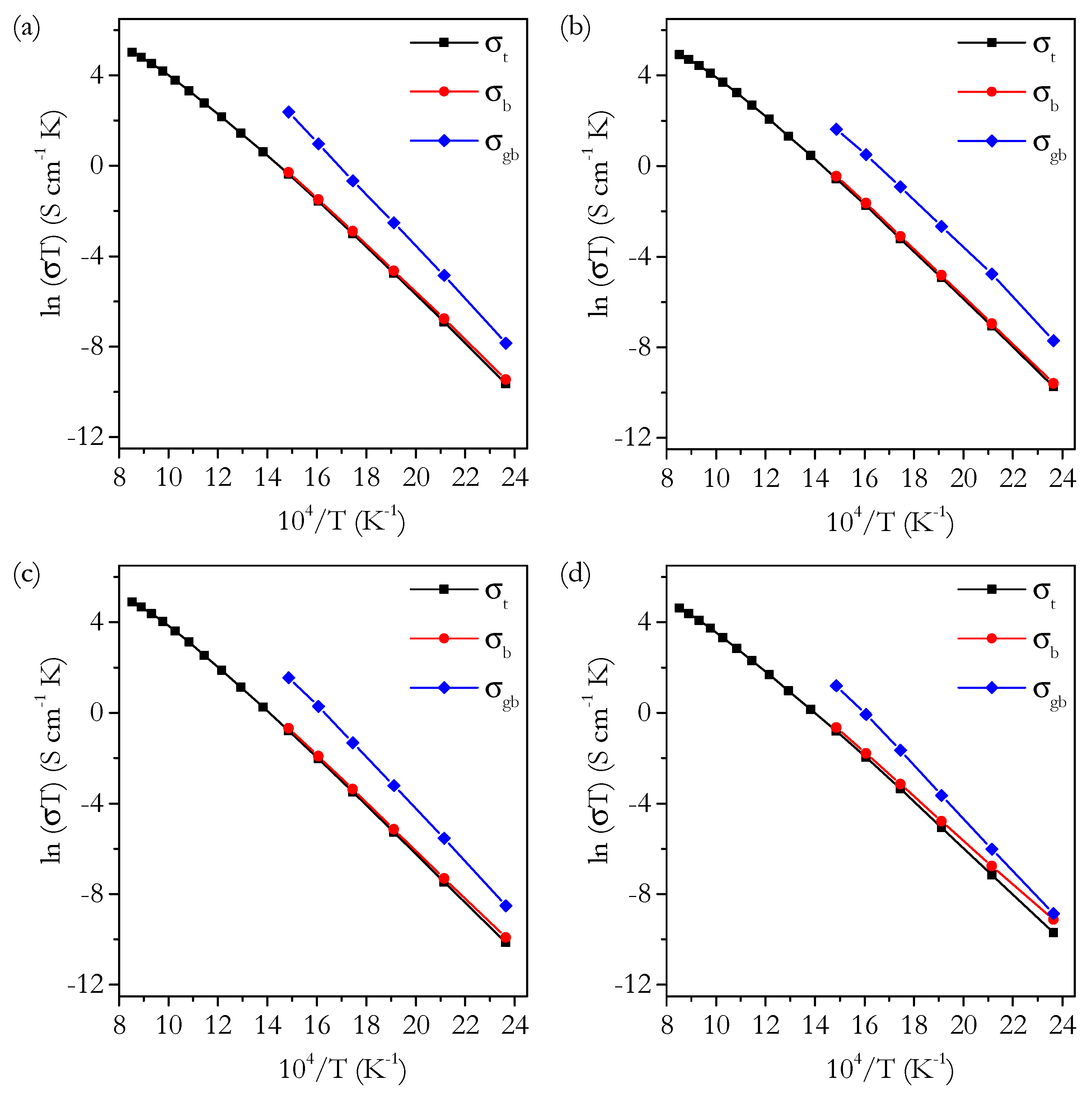
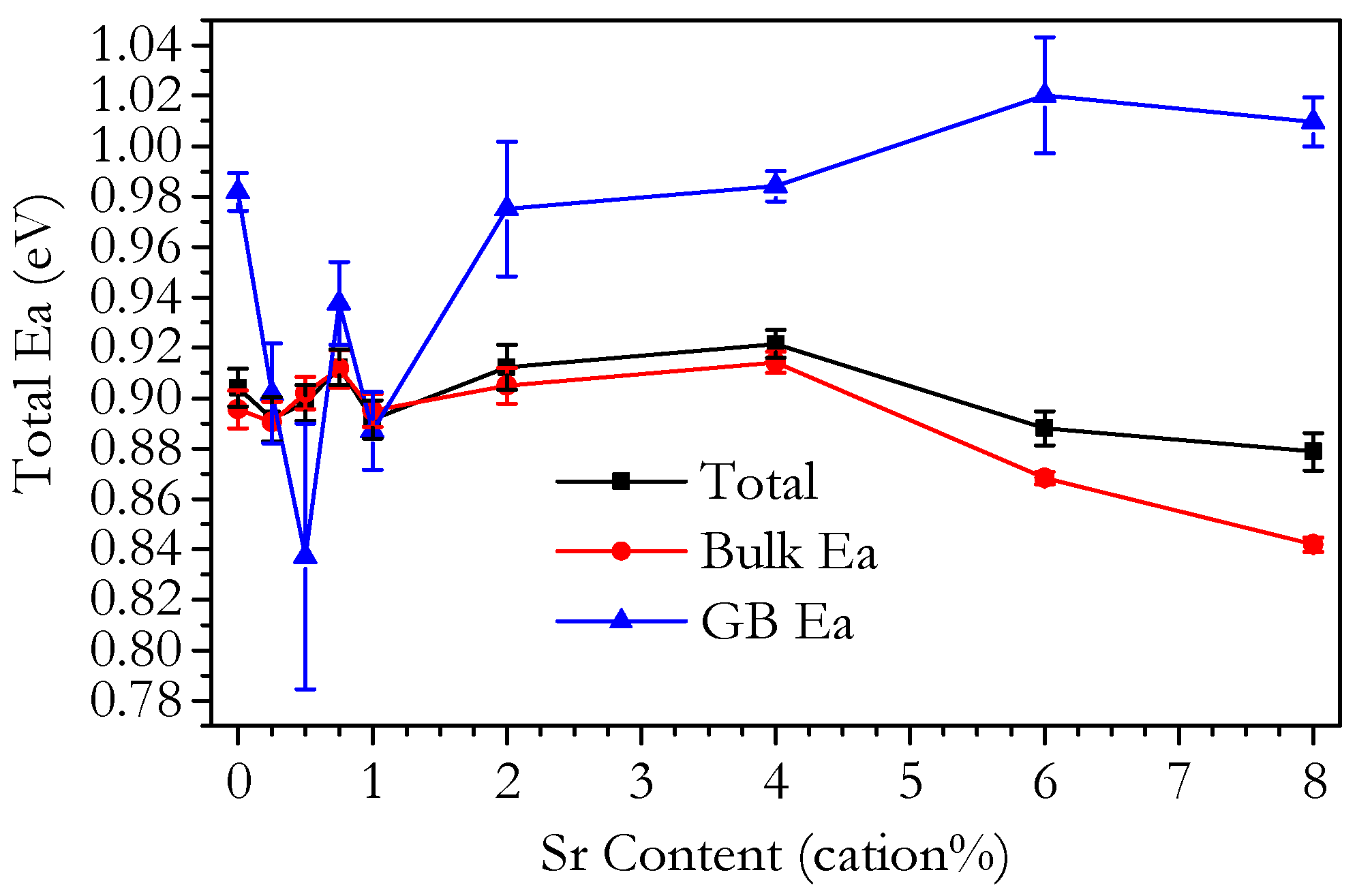
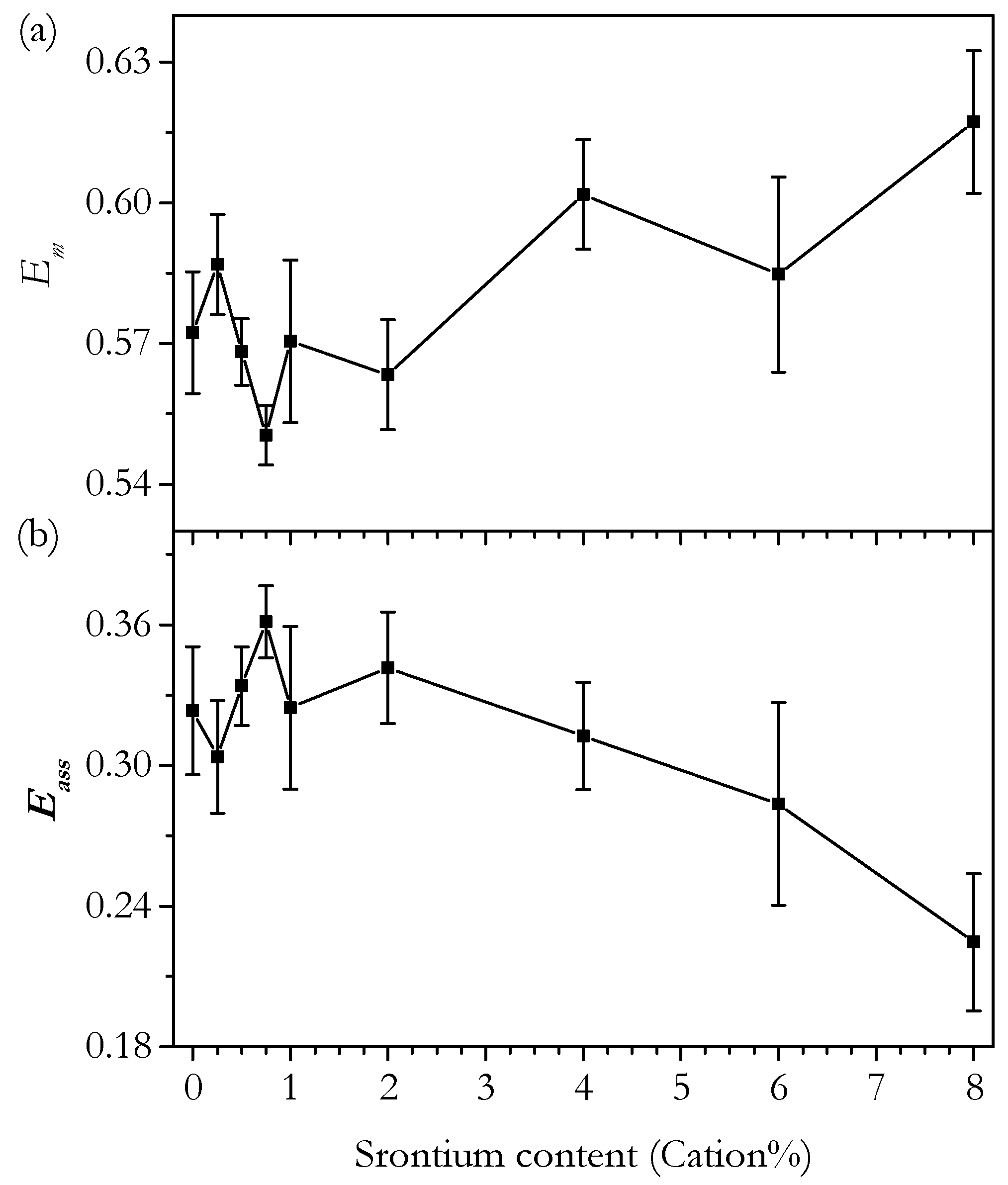
3.3. Conductivity
3.3.1. Conductivity of Phase-Pure Samples
3.3.2. Conductivity of Bi-Phasic Sr6.00 and Sr8.00 Samples
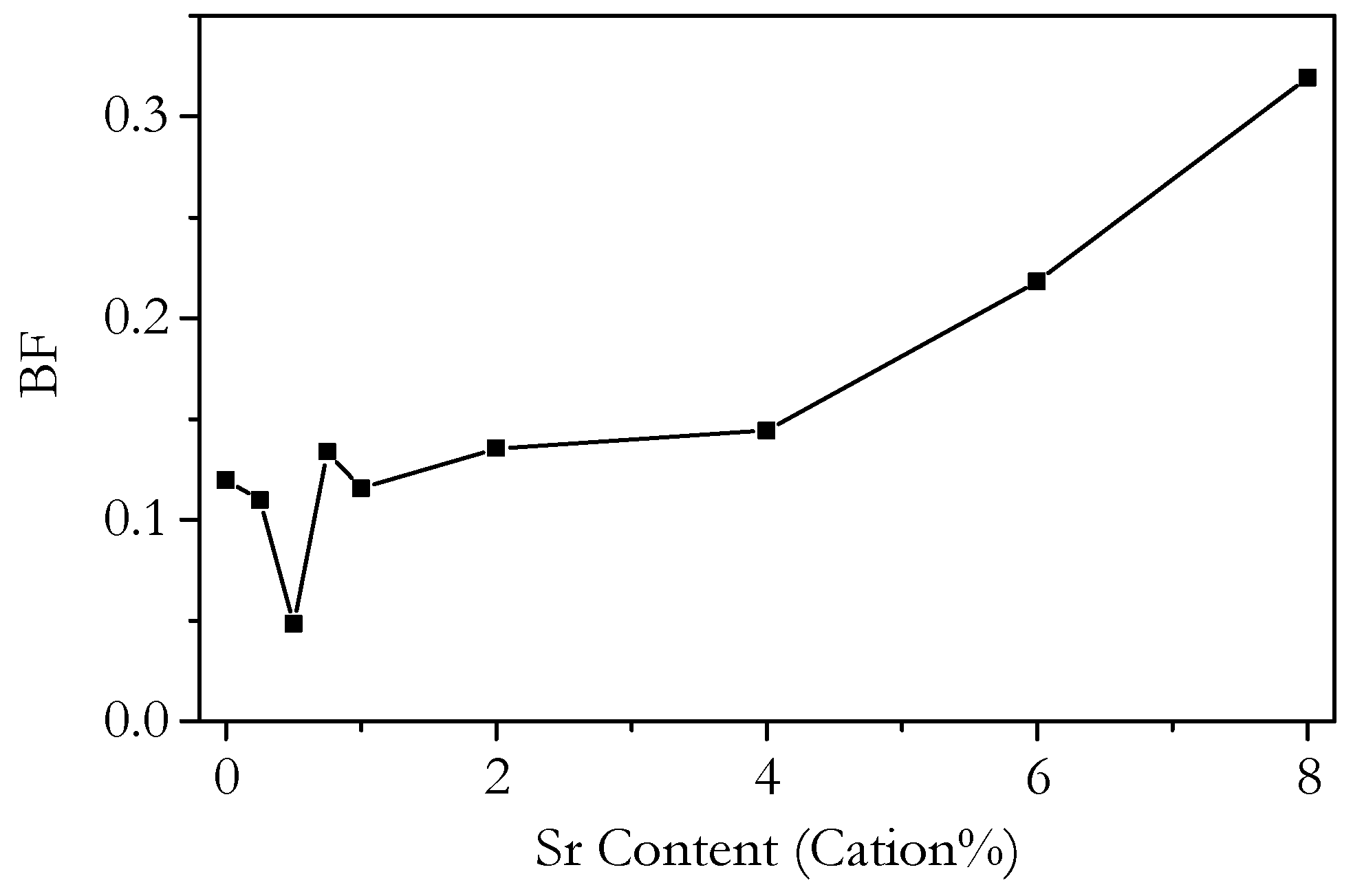
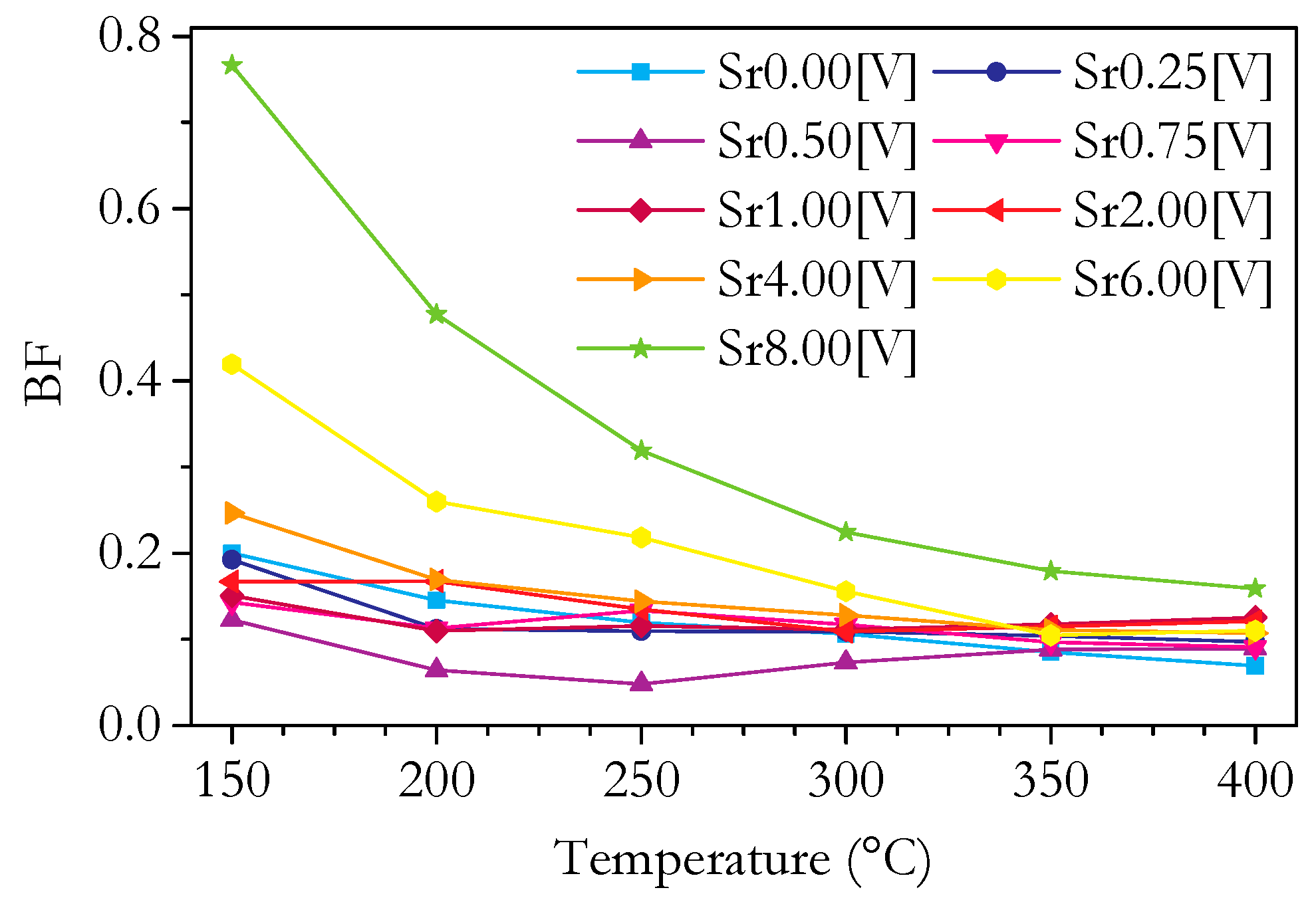
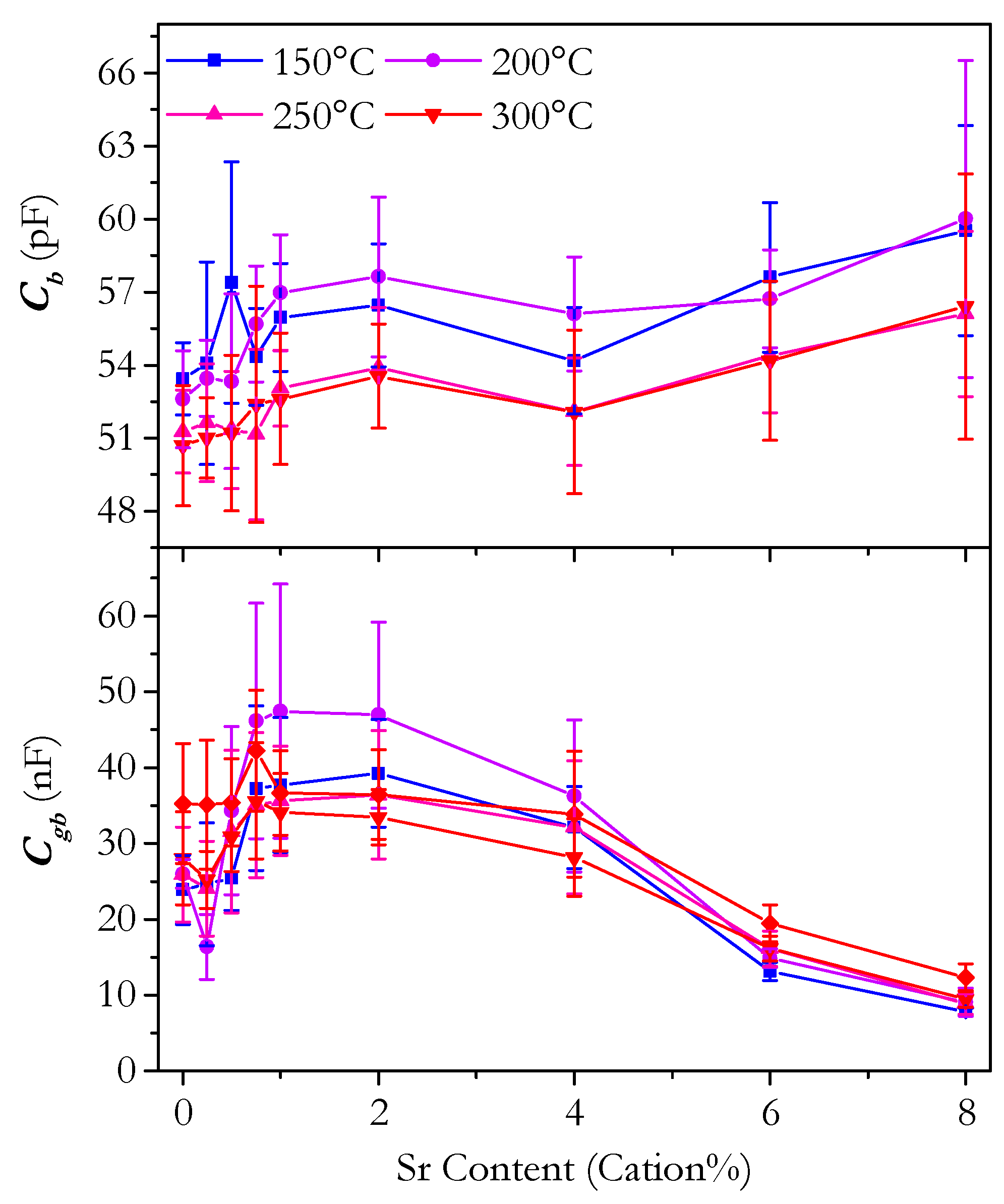
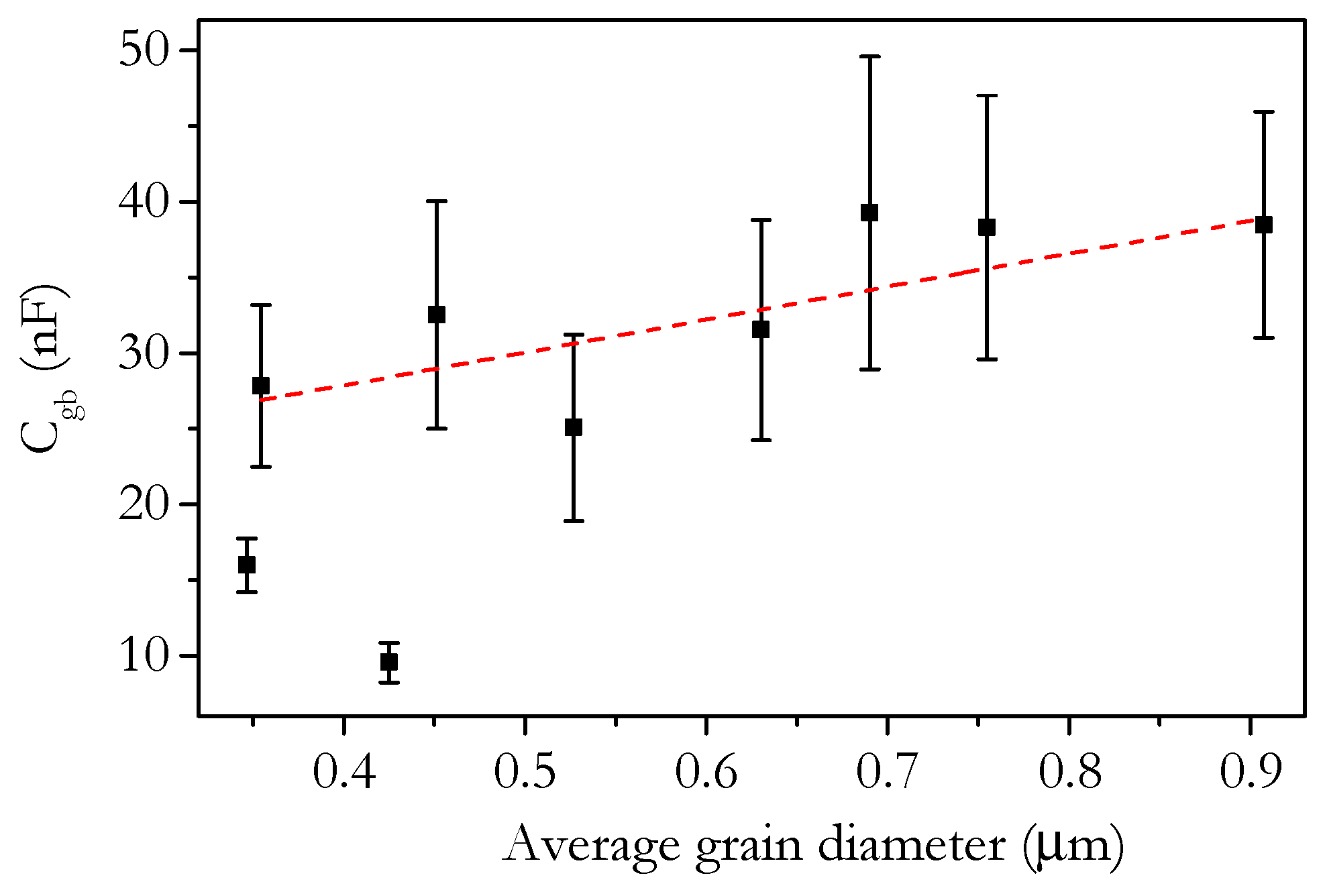
3.4. Capacitance
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minh, N.M.; Takahashi, T. Science and Technology of Ceramic Fuel Cells, 1st ed.; Elsevier: New York, NY, USA, 1995; pp. 1–14. [Google Scholar]
- Sharifzadeh, M. (Ed.) Design and Operation of Solid Oxide Fuel Cells: The Systems Engineering Vision; Elsevier: New York, NY, USA, 2019. [Google Scholar]
- Fergus, J.W.; Hui, R.; Li, X.; Wilkinson, D.P.; Zhang, J. (Eds.) Solid Oxide Fuel Cells: Materials Properties and Performance; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Huang, K.; Goodenough, J.B. Solid Oxide Fuel Cell Technology: Principles, Performance and Operations; Elsevier: New York, NY, USA, 2009. [Google Scholar]
- Wachsman, E.D.; Lee, K.T. Lowering the Temperature of Solid Oxide Fuel Cells. Science 2011, 334, 935–939. [Google Scholar] [CrossRef]
- Jacobson, A.J. Materials for Solid Oxide Fuel Cells. Chem. Mater. 2010, 22, 660–674. [Google Scholar] [CrossRef]
- Leah, R.; Bone, A.; Lankin, M.; Selcuk, A.; Pierce, R.; Rees, L.; Corcoran, D.; Muhl, P.; Dehaney-Steven, Z.; Brackenbury, C.; et al. Low-Cost, Redox-Stable, Low-Temperature SOFC Developed by Ceres Power for Multiple Applications: Latest Development Update. ECS Trans. 2013, 57, 461–470. [Google Scholar] [CrossRef]
- Kilner, J.A.; Burriel, M. Materials for Intermediate-Temperature Solid-Oxide Fuel Cells. Annu. Rev. Mater. Res. 2014, 44, 365–393. [Google Scholar] [CrossRef]
- Fuentes, R.O.; Baker, R.T. Structural and Electrochemical Properties of Gd0.1Ce0.9O1.95 Solid Solution Prepared by a Citrate Complexation Method. J. Power Sources 2009, 184, 268. [Google Scholar] [CrossRef]
- Kosinski, M.R.; Baker, R.T. Preparation and Property-Performance Relationships in Samarium-Doped Ceria Nanopowders for SOFC Electrolytes. J. Power Sources 2011, 196, 2498. [Google Scholar] [CrossRef]
- Coles-Aldridge, A.V.; Baker, R.T. Ionic Conductivity in Multiply Substituted Ceria-based Electrolytes. Solid State Ion. 2018, 316, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Coles-Aldridge, A.V.; Baker, R.T. Oxygen Ion conductivity in ceria-based electrolytes co-doped with samarium and gadolinium. Solid State Ion. 2020, 347, 115255. [Google Scholar] [CrossRef]
- Lane, J.; Neff, J.; Christie, G. Mitigation of the Deleterious Effect of Silicon Species on the Conductivity of Ceria Electrolytes. Solid State Ion. 2006, 177, 1911–1915. [Google Scholar] [CrossRef]
- Yeh, T.-H.; Chou, C.-C. Ionic Conductivity Investigation in Samarium and Strontium Co-doped Ceria System. Phys. Scr. 2007, T129, 303–307. [Google Scholar] [CrossRef]
- Kim, D.K.; Cho, P.S.; Lee, J.H.; Kim, D.Y. Mitigation of Highly Resistive Grain-boundary Phase in Gadolinia-doped Ceria by the Addition of SrO. Electrochem. Solid-State Lett. 2007, 10, 91–95. [Google Scholar] [CrossRef]
- Cioateră, N.; Pârvulescu, V.; Rolle, A.; Vannier, R.N. Effect of Strontium Addition on Europium-doped Ceria Properties. Solid State Ion. 2009, 180, 681–687. [Google Scholar] [CrossRef]
- Ramesh, S.; Reddy, C.V. Properties of Al2O3–Sm2O3–CeO2 Electrolyte. Acta Phys. Pol. A 2009, 115, 909–913. [Google Scholar] [CrossRef]
- Zheng, Y.; He, S.; Ge, L.; Zhou, M.; Chen, H.; Guo, L. Effect of Sr on Sm-doped Ceria Electrolyte. Int. J. Hydrogen Energy 2011, 36, 5128–5135. [Google Scholar] [CrossRef]
- Buchi Suresh, M.; Johnson, R. The Effect of Strontium Doping on Densification and Electrical Properties of Ce0.8Gd0.2O2−δ Electrolyte for IT-SOFC Application. Ionics (Kiel) 2011, 18, 291–297. [Google Scholar]
- Ramesh, S.; Raju, K.C.J.; Reddy, C.V. Synthesis and Characterization of Co-Doped Ceria Ceramics by Sol-Gel Method. Trans. Indian Ceram. Soc. 2011, 70, 143–147. [Google Scholar] [CrossRef]
- Siqueira, J.M., Jr.; Brum Malta, L.F.; Garrido, F.M.S.; Ogasawara, T.; Medeiros, M.E. Raman and Rietveld Structural Characterization of Sintered Alkaline Earth Doped Ceria. Mater. Chem. Phys. 2012, 135, 957–964. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, X.; Bergman, B.; Zhao, Z. Enhanced Ionic Conductivity of Ce0.8Sm0.2O2-δ by Sr Addition. J. Power Sources 2012, 208, 225–231. [Google Scholar] [CrossRef]
- Horovistiz, A.L.; Muccillo, E.N.S. Microstructural and Electrical Characterizations of Chemically Prepared Ce0.8Gd0.2− x(Ag, Sr)xO1.9 (0 ≤ x ≤ 0.02). Solid State Ion. 2012, 225, 428–431. [Google Scholar] [CrossRef]
- Jaiswal, N.; Kumar, D.; Upadhyay, S.; Parkash, O. Effect of Mg and Sr Co-doping on the Electrical Properties of Ceria-based Electrolyte Materials for Intermediate Temperature Solid Oxide Fuel Cells. J. Alloys Compd. 2013, 577, 456–462. [Google Scholar] [CrossRef]
- Jaiswal, N.; Upadhyay, S.; Kumar, D.; Parkash, O. Sm3+ and Sr2+ Co-doped Ceria Prepared by Citrate–Nitrate Auto-combustion Method. Int. J. Hydrogen Energy 2014, 39, 543–551. [Google Scholar] [CrossRef]
- Kashyap, D.; Patro, P.K.; Lenka, R.K.; Mahata, T.; Sinha, P.K. Effects of Gd and Sr Co-doping in CeO2 for Electrolyte Application in Solid Oxide Fuel Cell (SOFC). Ceram. Int. 2014, 40, 11869–11875. [Google Scholar] [CrossRef]
- Sherwood, T.; Baker, R.T. Effects of Strontium Content on the Microstructure and Ionic Conductivity of Samarium-Doped Ceria. Solids 2021, 2, 293–313. [Google Scholar] [CrossRef]
- Fergus, J.W. Electrolyte for Solid Oxide Fuel Cells. J. Power Sources 2006, 162, 30–40. [Google Scholar] [CrossRef]
- Klug, H.; Alexander, L. X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials; John Wiley: New York, NY, USA, 1974. [Google Scholar]
- Yahiro, H.; Eguchi, Y.; Eguchi, K.; Arai, H. Oxygen Ion Conductivity of the Ceria-Samarium Oxide System with Fluorite Structure. J. Appl. Electrochem. 1988, 18, 527–531. [Google Scholar] [CrossRef]
- Matovic, B.; Bucevac, D.; Jiraborvornpongsa, N.; Yoshida, K.; Yano, T. Synthesis and Characterization of Nanometric Strontium-doped Ceria Solid Solutions via Glycine-Nitrate Procedure. J. Ceram. Soc. Japan 2012, 120, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, D. Effect of Y on the Properties of Sm-Doped Ceria for IT-SOFC Applications. J. Am. Ceram. Soc. 1989, 72, 1415–1421. [Google Scholar] [CrossRef]
- Hong, S.J.; Virkar, A.V. Lattice Parameters and Densities of Rare-Earth Oxide Doped Ceria Electrolytes. J. Am. Ceram. Soc. 1995, 78, 433–439. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Kümmerle, E.; Heger, G. The Structures of C–Ce2O3+δ, Ce7O12, and Ce11O20. J. Solid State Chem. 1999, 147, 485–500. [Google Scholar] [CrossRef]
- Jaiswal, N.; Singh, N.K.; Kumar, D.; Parkash, O. Effect of Strontium (Sr) Doping on the Conductivity of Ceria. J. Power Sources 2012, 202, 78–84. [Google Scholar] [CrossRef]
- Anjaneya, K.C.; Nayaka, G.P.; Manjanna, J.; Ashwin Kumar, V.M.; Govindaraj, G.; Ganesha, K.N. Investigation on the Sr-doped Ceria Ce1−xSrxO2−δ (x = 0.05–0.2) as an Electrolyte for Intermediate Temperature SOFC. J. Alloys Compd. 2014, 598, 33–40. [Google Scholar] [CrossRef]
- Blumenthal, R.; Garnier, J. The Electrical Conductivity and Thermodynamic Behavior of SrO-doped Nonstoichiometric Cerium Dioxide. J. Solid State Chem. 1976, 16, 21–34. [Google Scholar] [CrossRef]
- Cho, P.; Park, S.-Y.; Kim, J.; Do, H.; Park, H.; Lee, J.H. Diffusion Induced Grain-boundary Migration in SrO-doped CeO2 Electrolyte and its Effect on Electrical Properties. Solid State Ion. 2010, 181, 1420–1424. [Google Scholar] [CrossRef]
- Milliken, C.E.; Guruswamy, S.; Khandkar, A.C. Electrochemical Stability of Strontium-Doped Ceria Electrolyte in Solid-Oxide Fuel Cell Applications. J. Am. Ceram. Soc. 2004, 84, 1533–1538. [Google Scholar] [CrossRef]
- Yahiro, H.; Eguchi, K.; Arai, H. Ionic Conduction and Microstructure of the Ceria-Strontia System. Solid State Ion. 1986, 21, 37–47. [Google Scholar] [CrossRef]
- Longo, V.; Meriani, S.; Ricciardiello, F. Subsolidus Phase Relations Between 900° and 1700 °C in the Systems BeO-MgO-CeO2, SrO-MgO-CeO2, BaO-MgO-CeO2, and BaO-CaO-CeO2. J. Am. Ceram. Soc. 1981, 64, C-38–C-39. [Google Scholar] [CrossRef]
- Chavan, S.V.; Tyagi, A.K. Sub-solidus Phase Equilibria in CeO2–SrO System. Thermochim. Acta 2002, 390, 79–82. [Google Scholar] [CrossRef]
- Anjaneya, K.C.; Nayaka, G.P.; Manjanna, J.; Govindaraj, G.; Ganesha, K.N. Studies on Structural, Morphological and Electrical Properties of CeO.8Ln0.2O2−δ (Ln = Y3+, Gd3+, Sm3+, Nd3+ and La3+) Solid Solutions Prepared by Citrate Complexation Method. J. Alloys Compd. 2014, 585, 594–601. [Google Scholar] [CrossRef]
- Chen, P.-L.; Chen, I.-W. Grain Growth in CeO2: Dopant Effects, Defect Mechanism, and Solute Drag. J. Am. Ceram. Soc. 1996, 79, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Beschnitt, S.; Zacherle, T.; De Souza, R.A. Computational Study of Cation Diffusion in Ceria. J. Phys. Chem. C 2015, 119, 27307–27315. [Google Scholar] [CrossRef]
- Rahaman, M.N. Sintering of Ceramics, 1st ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 105–172. [Google Scholar]
- Inaba, H.; Tagawa, H. Ceria-based Solid Electrolytes. Solid State Ion. 1996, 83, 1–16. [Google Scholar] [CrossRef]
- Ruifeng, G.; Zongqiang, M. Sintering of Ce0.8Sm0.2O1.9. J. Rare Earths 2007, 25, 364–367. [Google Scholar] [CrossRef]
- Ding, D.; Liu, B.; Zhu, Z.; Zhou, S.; Xia, C. High Reactive Ce0.8Sm0.2O1.9 Powders via a Carbonate Co-precipitation Method as Electrolytes for Low-temperature Solid Oxide Fuel Cells. Solid State Ion. 2008, 179, 896–899. [Google Scholar] [CrossRef]
- Van Herle, J.; Seneviratne, D.; McEvoy, A.J. Lanthanide Co-doping of Solid Electrolytes: AC Conductivity Behaviour. J. Eur. Ceram. Soc. 1999, 19, 837–841. [Google Scholar] [CrossRef]
- Kim, J.; Lee, D. The Effect of Multiple Doping on Electrical Conductivity of Gadolinia-doped Ceria Electrolyte. Korean J. Chem. Eng. 2002, 19, 421–424. [Google Scholar] [CrossRef]
- Wang, F.-Y.; Chen, S.; Cheng, S. Gd3+ and Sm3+ Co-doped Ceria Based Electrolytes for Intermediate Temperature Solid Oxide Fuel Cells. Electrochem. Commun. 2004, 6, 743–746. [Google Scholar] [CrossRef]
- Omar, S.; Wachsman, E.D.; Nino, J.C. Higher Ionic Conductive Ceria-based Electrolytes for Solid Oxide Fuel Cells. Appl. Phys. Lett. 2007, 91, 144106. [Google Scholar] [CrossRef]
- Liu, Y.; Li, B.; Wei, X.; Pan, W. Citric–Nitrate Combustion Synthesis and Electrical Conductivity of the Sm3+ and Nd3+ Co-Doped Ceria Electrolyte. J. Am. Ceram. Soc. 2008, 91, 3926–3930. [Google Scholar] [CrossRef]
- Guan, X.; Zhou, H.; Liu, Z.; Wang, Y.; Zhang, J. Preparation and Properties of Gd3+ and Y3+ Co-doped Ceria-based Electrolytes for Intermediate Temperature Solid Oxide Fuel Cells. J. Alloys Compd. 2008, 464, 310–316. [Google Scholar] [CrossRef]
- Li, B.; Wei, X.; Pan, W. Improved Electrical Conductivity of Ce0.9Gd0.1O1.95 and Ce0.9Sm0.1O1.95 by Co-doping. Int. J. Hydrogen Energy 2010, 35, 3018–3022. [Google Scholar] [CrossRef]
- Li, B.; Liu, Y.; Wei, X.; Pan, W. Electrical Properties of Ceria Co-doped with Sm3+ and Nd3+. J. Power Sources 2010, 195, 969–976. [Google Scholar] [CrossRef]
- Yao, H.-C.; Zhang, Y.-X.; Liu, J.-J.; Li, Y.-L.; Wang, J.-S.; Li, Z.-J. Synthesis and Characterization of Gd3+ and Nd3+ Co-doped Ceria by using Citric Acid–Nitrate Combustion Method. Mater. Res. Bull. 2011, 46, 75–80. [Google Scholar] [CrossRef]
- Kasse, R.M.; Nino, J.C. Ionic Conductivity of SmxNdyCe0.9O2−δ Co-doped Ceria Electrolytes. J. Alloys Compd. 2013, 575, 399–402. [Google Scholar] [CrossRef]
- Omar, S.; Wachsman, E.D.; Jones, J.L.; Nino, J.C. Crystal Structure–Ionic Conductivity Relationships in Doped Ceria Systems. J. Am. Ceram. Soc. 2009, 92, 2674–2681. [Google Scholar] [CrossRef]
- Mogensen, M.; Sammes, N.; Tompsett, G. Physical, Chemical and Electrochemical Properties of Pure and Doped Ceria. Solid State Ion. 2000, 129, 63–94. [Google Scholar] [CrossRef]
- Zhang, T.S.; Ma, J.; Chan, S.H.; Hing, P.; Kilner, J.A. Intermediate-temperature Ionic Conductivity of Ceria-based Solid Solutions as a Function of Gadolinia and Silica Contents. Solid State Sci. 2004, 6, 565–572. [Google Scholar] [CrossRef]
- Esposito, V.; Traversa, E. Design of Electroceramics for Solid Oxides Fuel Cell Applications: Playing with Ceria. J. Am. Ceram. Soc. 2008, 91, 1037–1051. [Google Scholar] [CrossRef]
- Ding, D.; Liu, B.; Gong, M.; Liu, X.; Xia, C. Electrical Properties of Samarium-doped Ceria Electrolytes of Highly Active Powders. Electrochim. Acta 2010, 55, 4529–4535. [Google Scholar] [CrossRef]
- Zhan, Z.; Wen, T.-L.; Tu, H.; Lu, Z.-Y. AC Impedance Investigation of Samarium-doped Ceria. J. Electrochem. Soc. 2001, 148, A427. [Google Scholar] [CrossRef]
- Irvine, J.T.S.; Sinclair, D.C.; West, A.R. Electroceramics: Characterization by Impedance Spectroscopy. Adv. Mater. 1990, 2, 132–138. [Google Scholar] [CrossRef]
- Christie, G.M.; van Berkel, F.P.F. Microstructure-Ionic Conductivity Relationships in Ceria-Gadolinia Electrolytes. Solid State Ion. 1996, 83, 17–27. [Google Scholar] [CrossRef]
- Verkerk, M.J.; Middelhuis, B.J.; Burggraaf, A.J. Effect of Grain Boundaries on the Conductivity of High-purity ZrO2/Y2O3 ceramics. Solid State Ion. 1982, 6, 159–170. [Google Scholar] [CrossRef] [Green Version]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Cation Concentration (Cation%) | |||||
|---|---|---|---|---|---|
| Sample Name | Ce | Sm | Sr | Gd | Total Minor Lanthanides |
| Sr0.00 | 79.2 ± 1.1 | 20.3 ± 0.3 | −0.05 ± 0.0002 | 0.476 ± 0.005 | 0.103 |
| Sr0.25 | 79.7 ± 0.4 | 19.5 ± 0.1 | 0.26 ± 0.01 | 0.473 ± 0.005 | 0.099 |
| Sr0.50 | 79.8 ± 1.5 | 19.1 ± 0.3 | 0.51 ± 0.01 | 0.475 ± 0.007 | 0.105 |
| Sr0.75 | 80.1 ± 0.6 | 18.5 ± 0.2 | 0.74 ± 0.02 | 0.462 ± 0.02 | 0.102 |
| Sr1.00 | 80.4 ± 0.7 | 18.0 ± 0.1 | 1.00 ± 0.01 | 0.483 ± 0.007 | 0.103 |
| Sr2.00 | 81.3 ± 0.4 | 16.1 ± 0.1 | 1.99 ± 0.02 | 0.483 ± 0.005 | 0.098 |
| Sr4.00 | 83.4 ± 0.4 | 12.0 ± 0.1 | 3.99 ± 0.03 | 0.490 ± 0.006 | 0.088 |
| Sr6.00 | 85.5 ± 0.6 | 8.0 ± 0.1 | 5.99 ± 0.07 | 0.494 ± 0.01 | 0.078 |
| Sr8.00 | 87.4 ± 0.5 | 4.0 ± 0.1 | 8.00 ± 0.1 | 0.514 ± 0.003 | 0.074 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherwood, T.; Baker, R.T. Effects on Microstructure and Ionic Conductivity of the Co-Doping with Strontium and Samarium of Ceria with Constant Oxygen Vacancy Concentration. Solids 2021, 2, 341-370. https://doi.org/10.3390/solids2040022
Sherwood T, Baker RT. Effects on Microstructure and Ionic Conductivity of the Co-Doping with Strontium and Samarium of Ceria with Constant Oxygen Vacancy Concentration. Solids. 2021; 2(4):341-370. https://doi.org/10.3390/solids2040022
Chicago/Turabian StyleSherwood, Toby, and Richard T. Baker. 2021. "Effects on Microstructure and Ionic Conductivity of the Co-Doping with Strontium and Samarium of Ceria with Constant Oxygen Vacancy Concentration" Solids 2, no. 4: 341-370. https://doi.org/10.3390/solids2040022
APA StyleSherwood, T., & Baker, R. T. (2021). Effects on Microstructure and Ionic Conductivity of the Co-Doping with Strontium and Samarium of Ceria with Constant Oxygen Vacancy Concentration. Solids, 2(4), 341-370. https://doi.org/10.3390/solids2040022