
A Brief History of Photoactive Interlocked Systems Assembled by Transition Metal Template Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
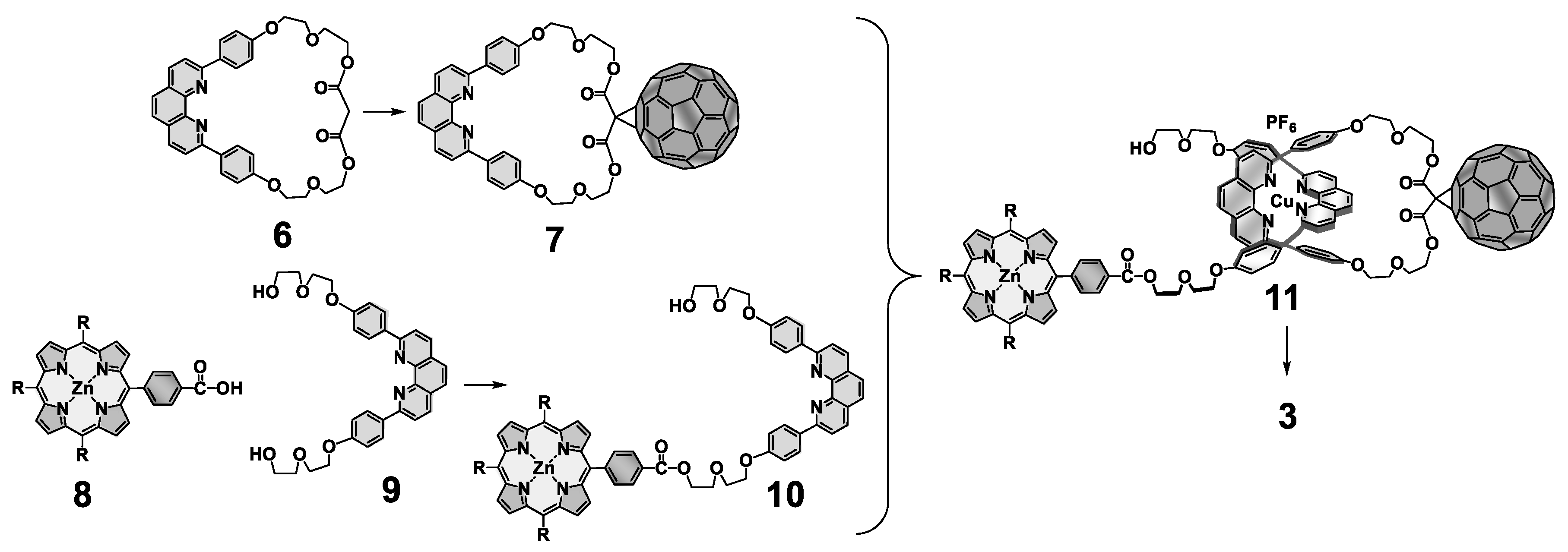{kind=link}
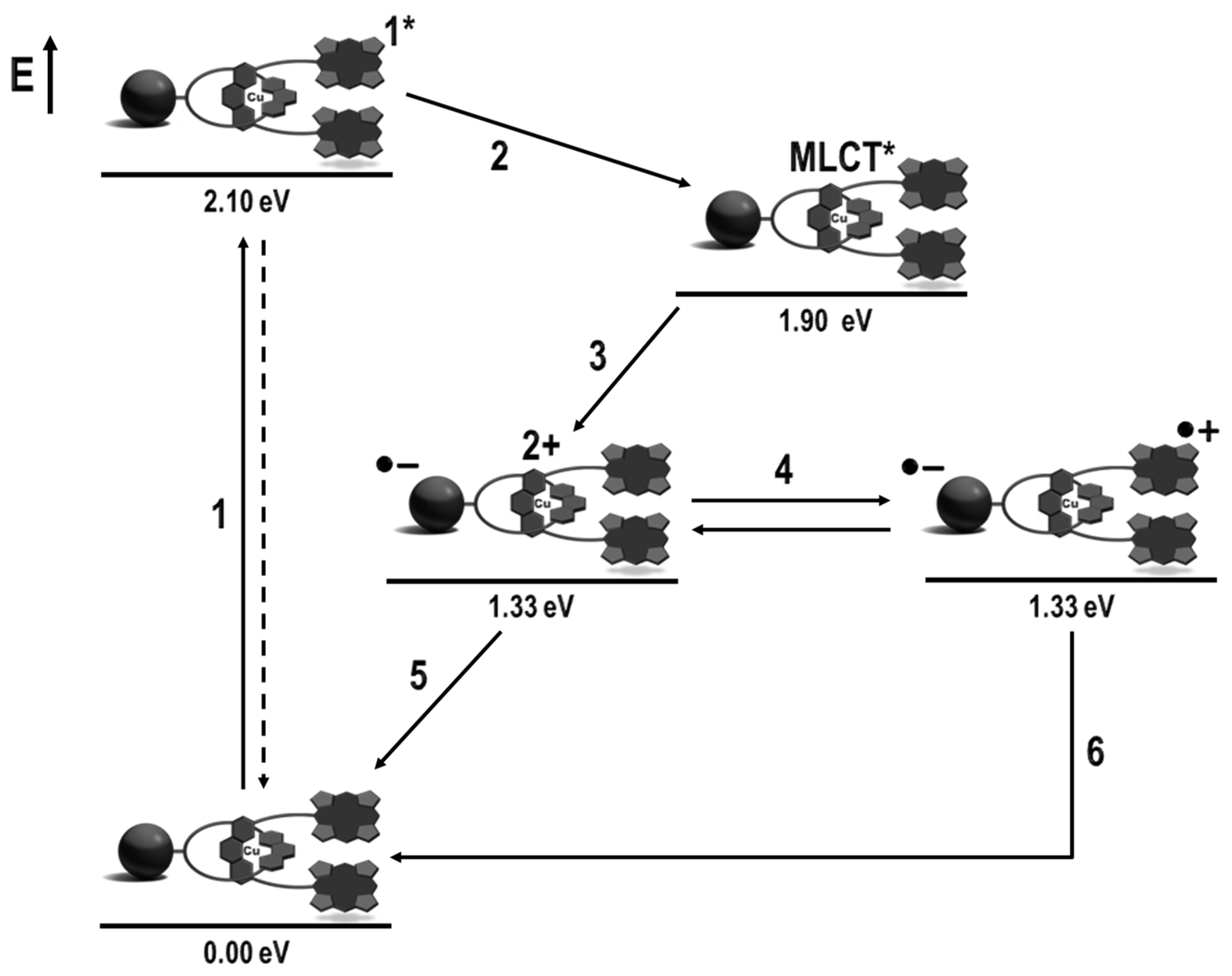{kind=link}
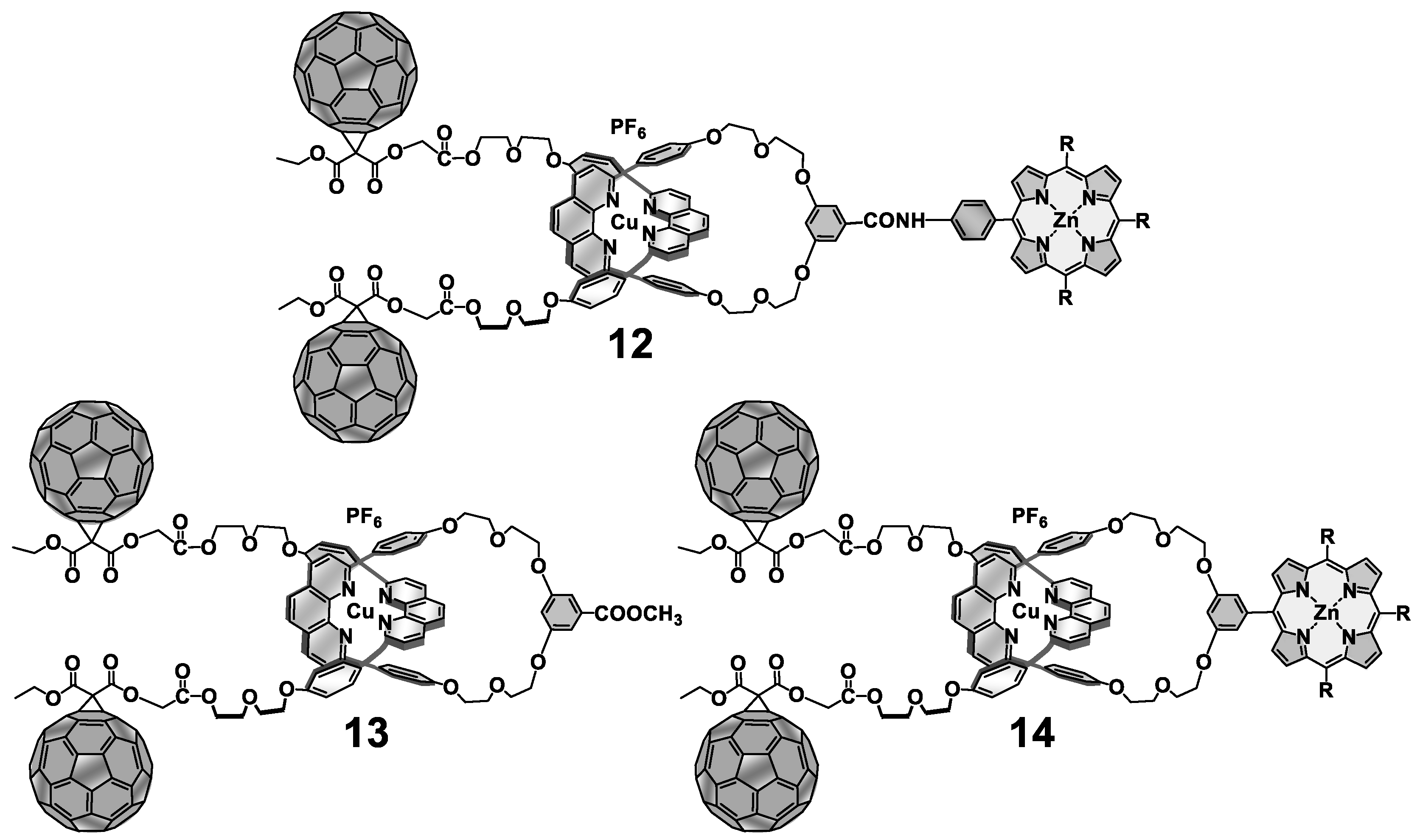{kind=link}
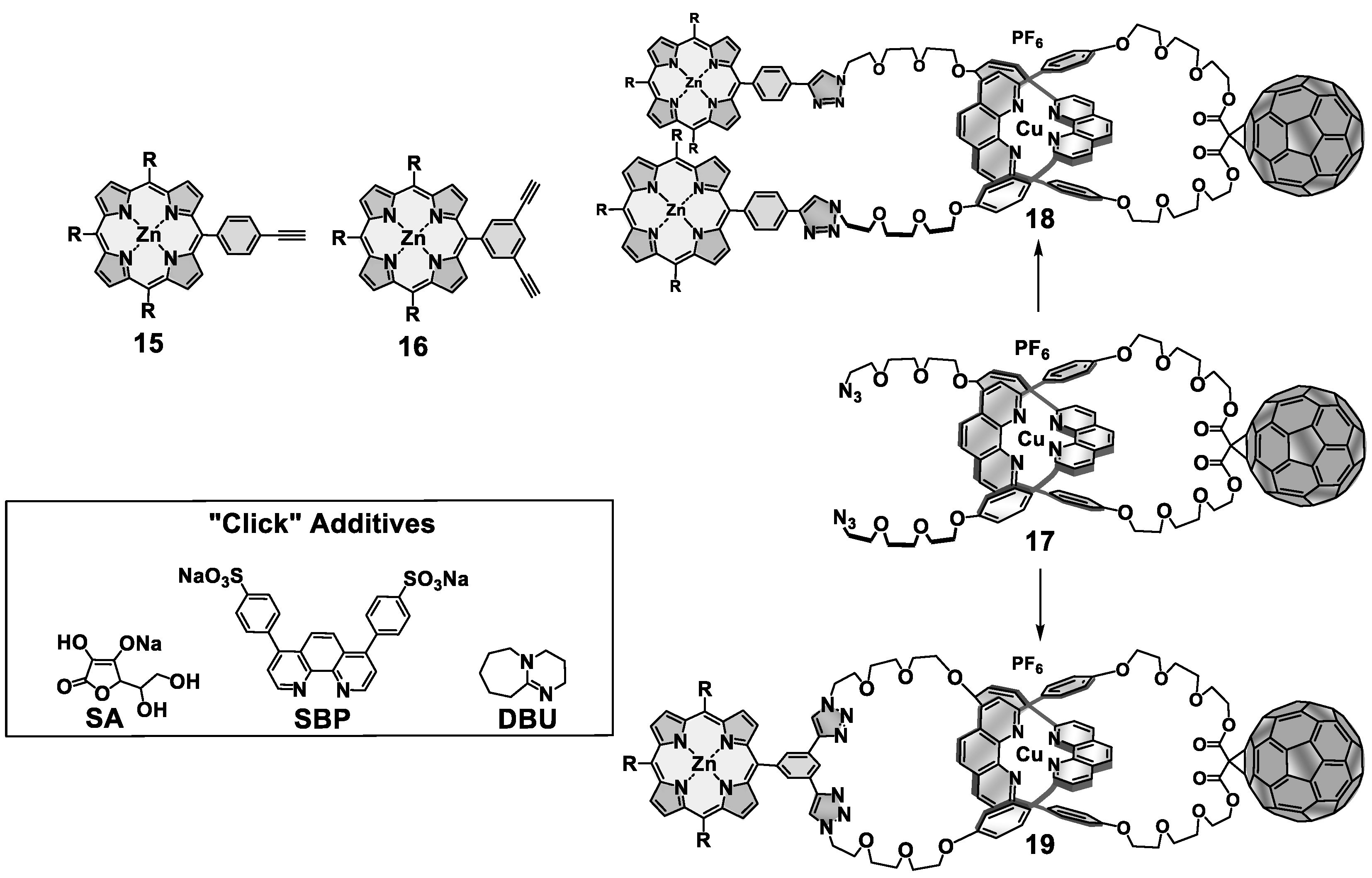{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
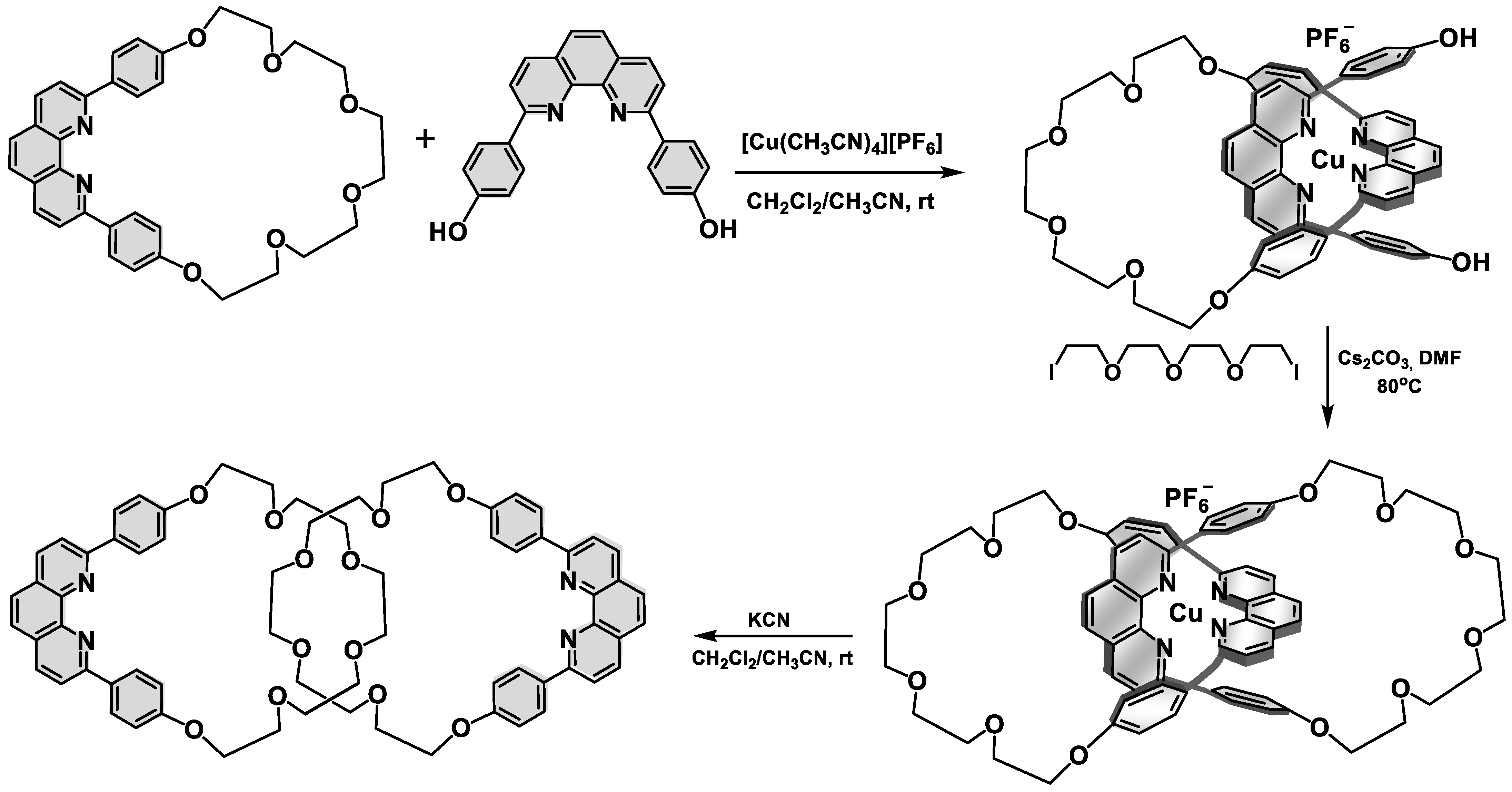
2. Interlocked Photosynthetic Models Decorated with Porphyrins as Electron Donors and Acceptors
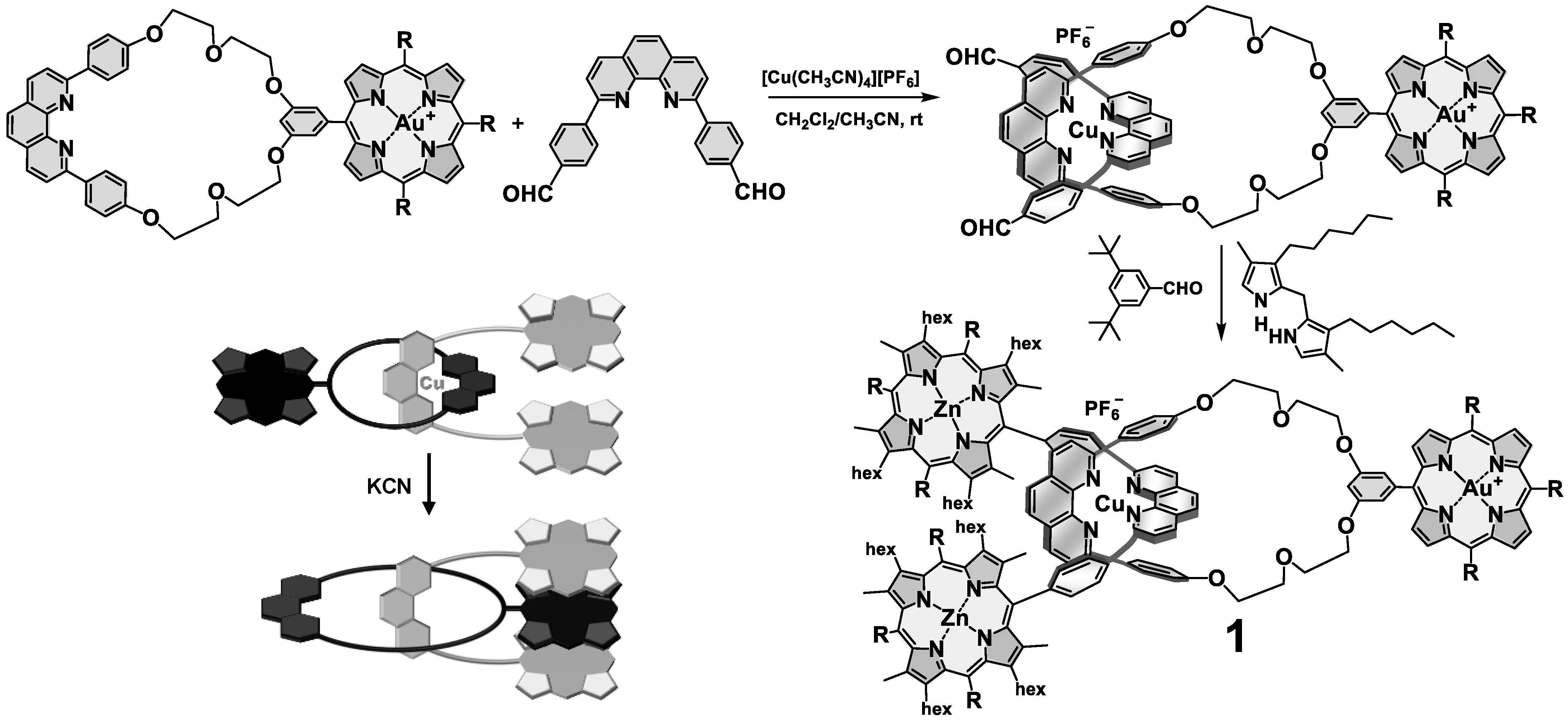
3. Interlocked Photosynthetic Models Decorated with Porphyrins as Electron Donors and Fullerenes as Acceptors
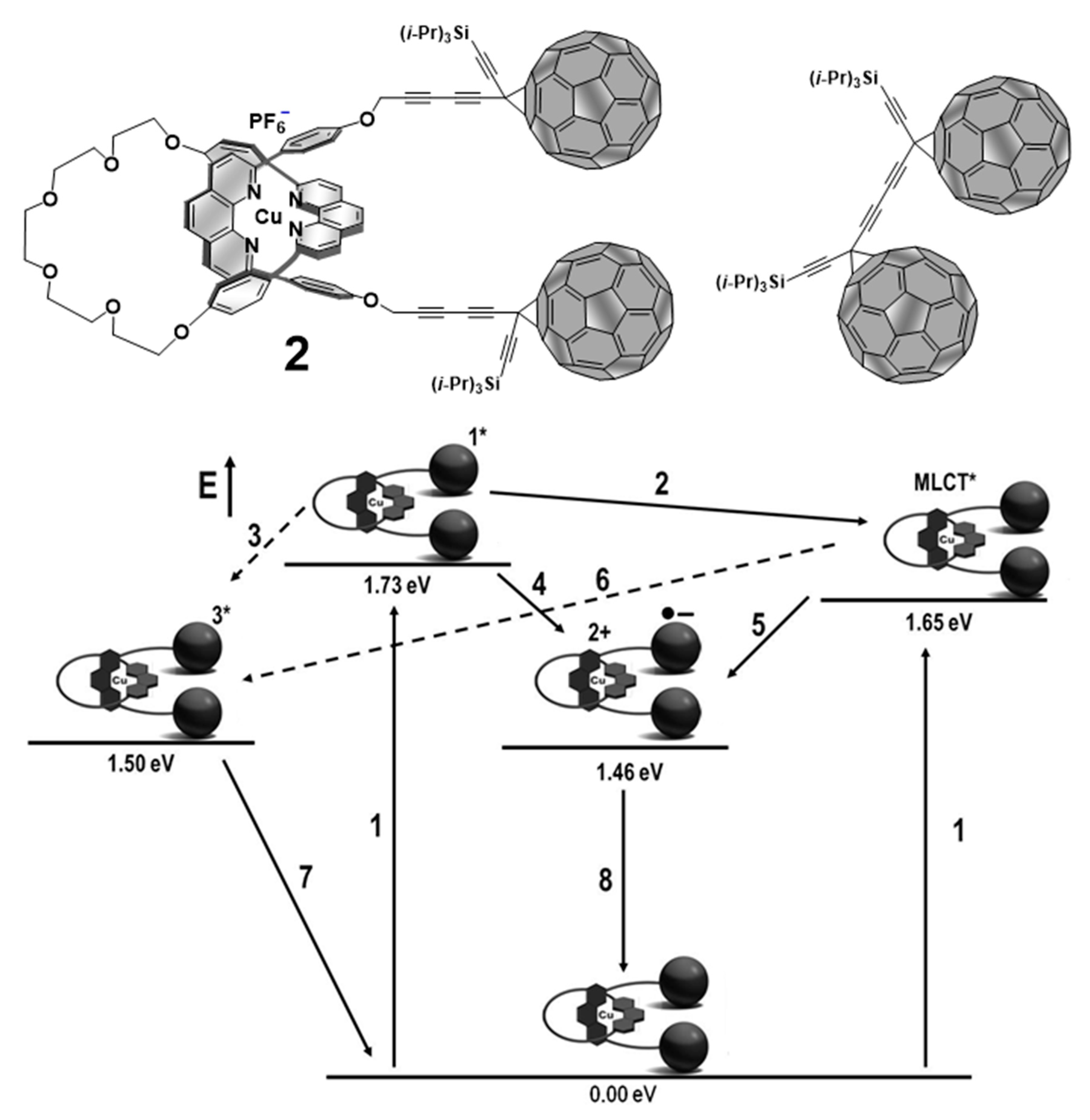
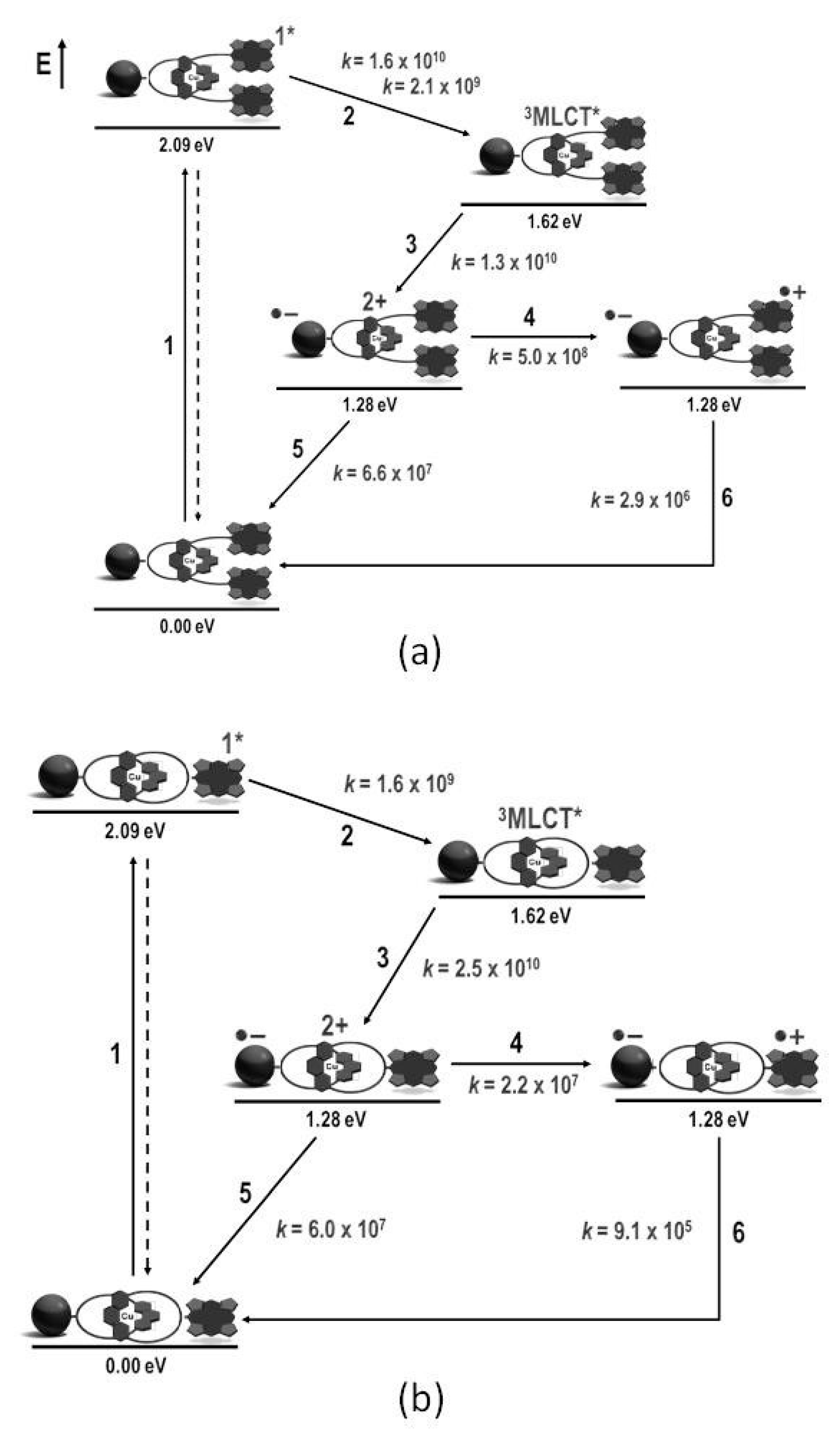
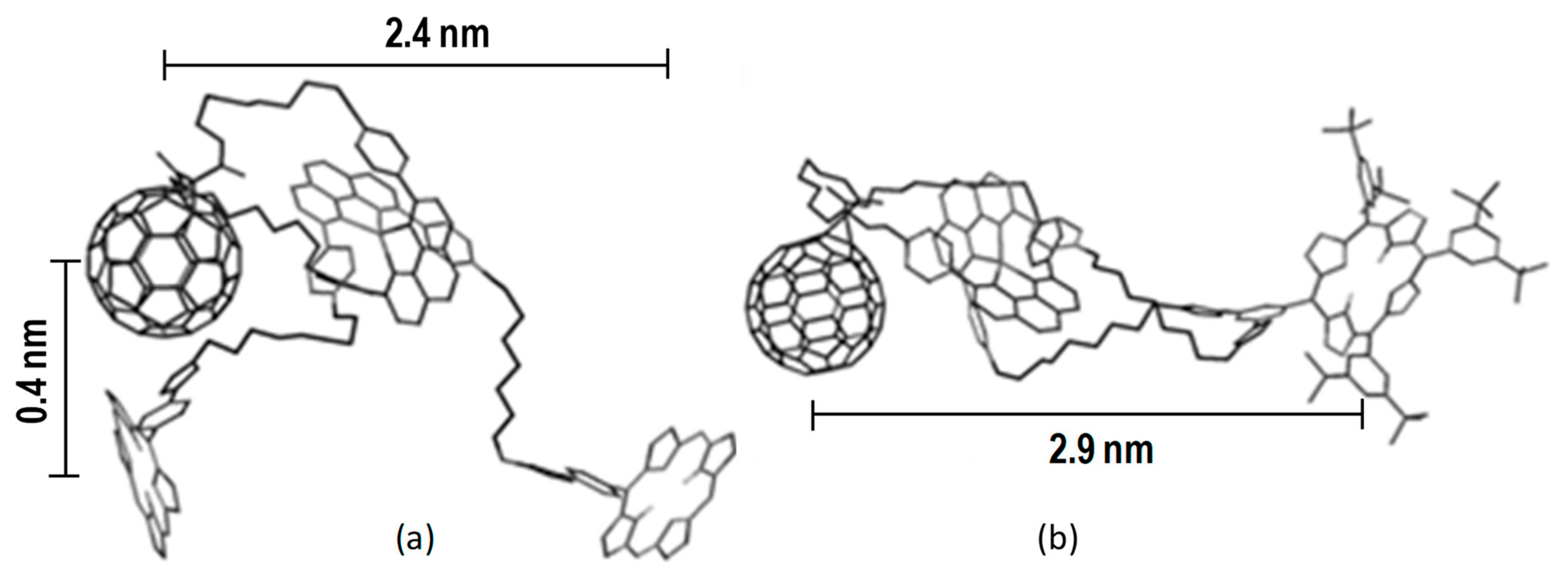
4. Effects of Molecular Topology on the Kinetics of Photoinduced Processes in Interlocked Molecules Decorated with Porphyrins as Electron Donors and Fullerenes as Acceptors
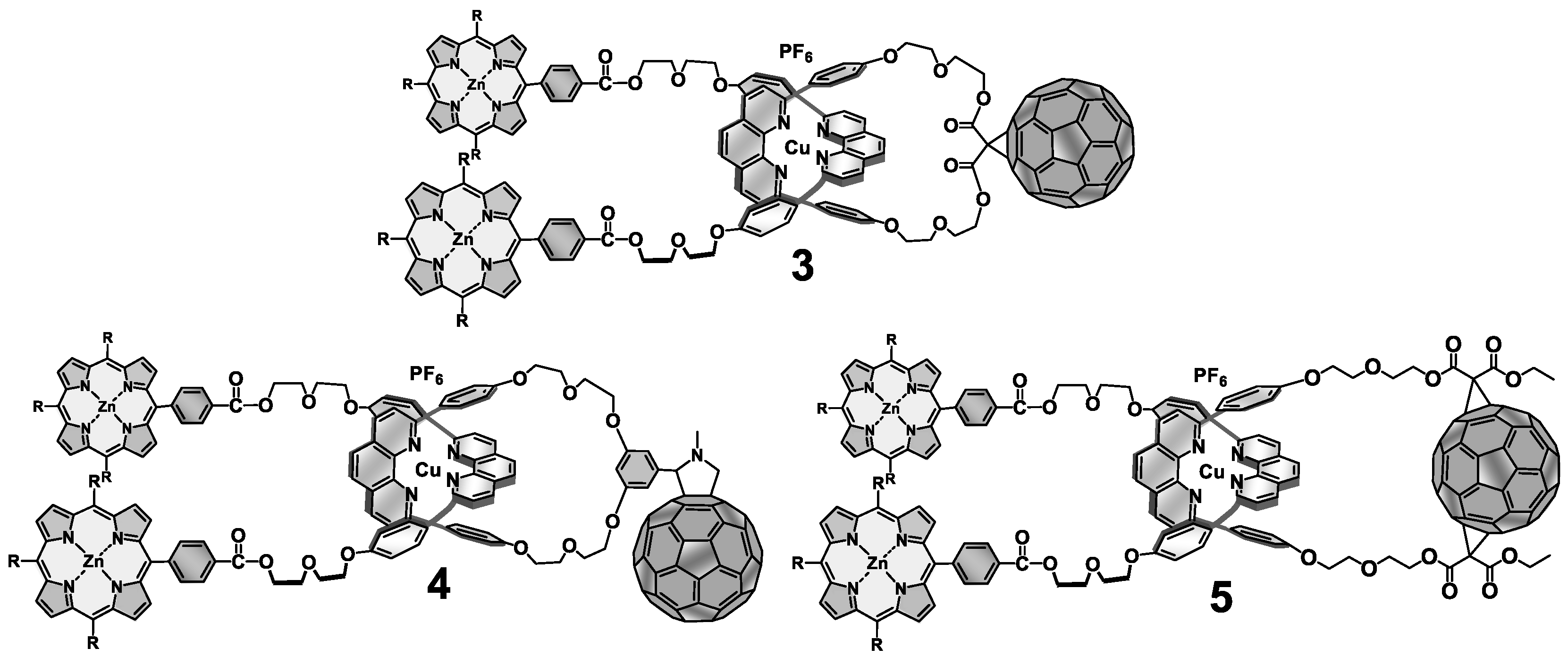
5. Multi-Chromophoric Rotaxanes as Artificial Photosynthetic Models
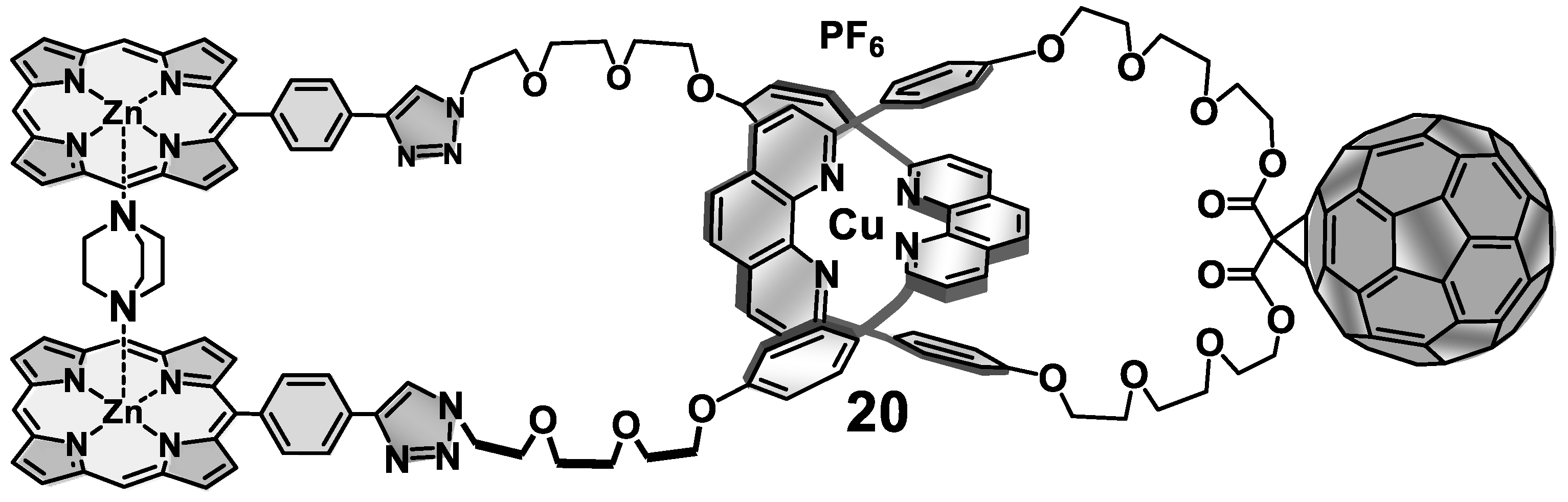
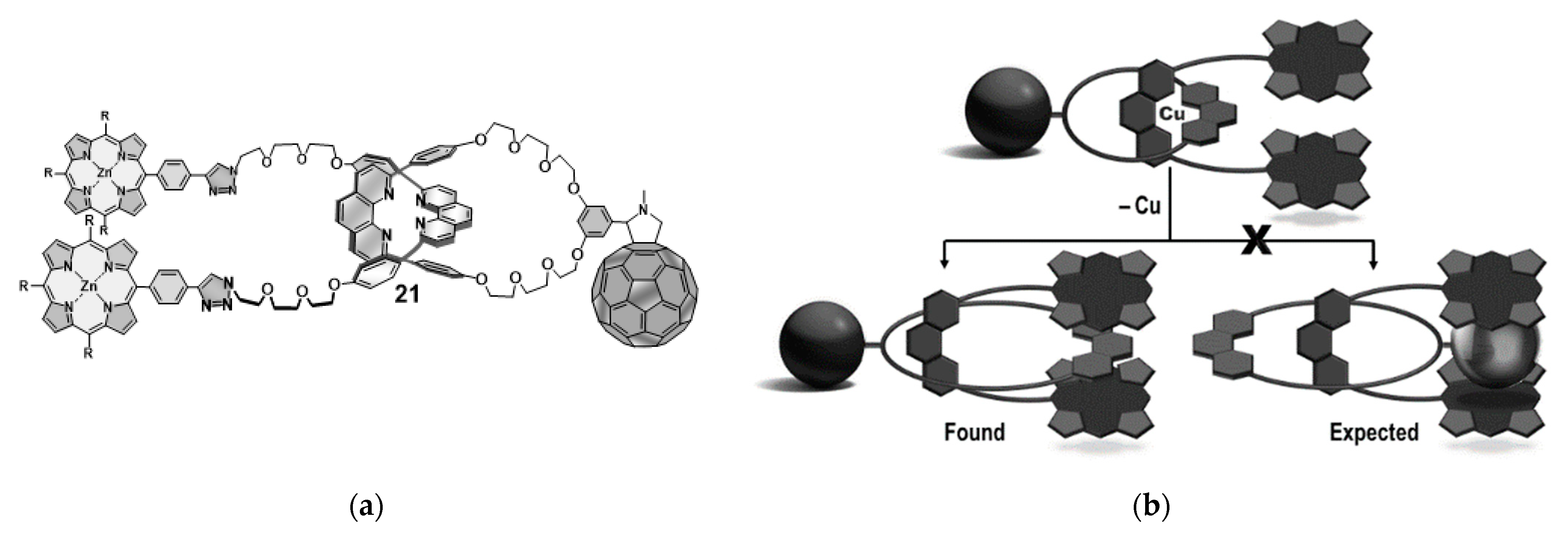
6. Active-Metal Template Synthesis of Photoactive Rotaxanes
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Burns, C.J.; Stoddart, J.F. The Nature of the Mechanical Bond: From Molecules to Machines; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Stoddart, J.F. Mechanically Interlocked Molecules (MIMs)-Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11094–11125. [Google Scholar] [CrossRef]
- Stoddart, J.F. The chemistry of the mechanical bond. Chem. Soc. Rev. 2009, 38, 1802–1820. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.-P. From Chemical Topology to Molecular Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11080–11093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, J.-P. Interlacing molecular threads on transition metals: Catenands, catenates, and knots. Acc. Chem. Res. 1990, 23, 319–327. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 18, 10081–10206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassem, S.; van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial Molecular Motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich-Buchecker, C.O.; Sauvage, J.-P. Interlocking of molecular threads: From the statistical approach to the templated synthesis of catenands. Chem. Rev. 1987, 87, 795–810. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives, 1st ed.; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Raymo, F.M.; Stoddart, J.F. Templated synthesis of catenanes and rotaxanes. In Templated Organic Synthesis; Diederich, F., Stang, P.J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000; pp. 75–104. [Google Scholar]
- Fujita, M. Metal-directed self-assembly of two- and three-dimensional synthetic receptors. Chem. Soc. Rev. 1998, 27, 417–425. [Google Scholar] [CrossRef]
- Vögtle, F.; Dünnwald, T.; Schmidt, T. Catenanes and Rotaxanes of the Amide Type. Acc. Chem. Res. 1996, 29, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Crowley, J.D.; Goldup, S.M.; Lee, A.-L.; Leigh, D.A.; McBurney, R.T. Active metal template synthesis of rotaxanes, catenanes and molecular shuttles. Chem. Soc. Rev. 2009, 38, 1530–1541. [Google Scholar] [CrossRef] [Green Version]
- Schalley, C.A.; Vögtle, F.; Dötz, K.H. Templates in Chemistry; Springer: Berlin, Germany, 2004; Volumes 1 and 2. [Google Scholar]
- Roche, C.; Sour, A.; Sauvage, J.-P. A Flexible Copper(I)-Complexed [4]Rotaxane Containing Two Face-to-Face Porphyrinic Plates that Behaves as a Distensible Receptor. Chem. Eur. J. 2012, 18, 8366–8376. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Bernt, I. Ligand and metal controlled multicomponent self-assembly of oligonuclear two- and three-dimensional complex molecular architectures. Curr. Opin. Solid State Mater. Sci. 1998, 3, 407–413. [Google Scholar] [CrossRef]
- Saatfrank, R.W.; Bernt, I.; Uller, E.; Hampel, F. Template-mediated self- assembly of six- and eight membered iron coronetes. Angew. Chem. Int. Ed. 1997, 36, 2482–2495. [Google Scholar] [CrossRef]
- Sauvage, J.P. Transition Metals in Supramolecular Chemistry; John Wiley & Sons, Inc.: Chichester, UK, 2008; Volume 5. [Google Scholar]
- Dietrich-Buchecker, C.O.; Sauvage, J.P.; Kintzinger, J.P. New family of molecules: The metallo-catenanes. Tetrahedron Lett. 1983, 24, 5095–5098. [Google Scholar] [CrossRef]
- Beves, J.E.; Blight, B.A.; Campbell, C.J.; Leigh, D.A.; McBurney, R.T. Strategies and Tactics for the Metal-Directed Synthesis of Rotaxanes, Knots, Catenanes, and Higher Order Links. Angew. Chem. Int. Ed. 2011, 50, 9260–9327. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A. Stable mixed phenanthroline copper (i) complexes. Key building blocks for supramolecular coordination chemistry. Chem. Commun. 1997, 11, 999–1000. [Google Scholar] [CrossRef]
- Saha, M.L.; Neogi, S.; Schmittel, M. Dynamic heteroleptic metal-phenanthroline complexes: From structure to function. Dalton Trans. 2014, 43, 3815–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasielewski, M.R. Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 1992, 92, 435–461. [Google Scholar] [CrossRef]
- Balzani, V. Supramolecular Photochemistry; Springer: Heidelberg, Germany, 1987. [Google Scholar]
- Balzani, V. Supramolecular Photochemistry. Tetrahedron 1992, 48, 10443–10514. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Collin, J.-P.; Dalbavie, J.-O.; Dietrich-Buchecker, C.O.; Heitz, V.; Odobel, F.; Solladie, N.; Sauvage, J.-P. Rotaxanes and other transition metal-assembled porphyrin arrays for long-range photoinduced charge separation. Coord. Chem. Rev. 1998, 178–180 Pt 2, 1299–1312. [Google Scholar] [CrossRef]
- Benniston, A.C. Photo- and redox-active [2]rotaxanes and [2]catenanes. Chem. Soc. Rev. 1996, 25, 427–435. [Google Scholar] [CrossRef]
- Segura, J.L.; Martin, N. New concepts in tetrathiafulvalene chemistry. Angew. Chem. Int. Ed. 2001, 40, 1372–1409. [Google Scholar] [CrossRef]
- Armaroli, N. Photoactive mono- and polynuclear Cu(I)-phenanthrolines. A viable alternative to Ru (II)-polypyridines? Chem. Soc. Rev. 2001, 30, 113–124. [Google Scholar] [CrossRef]
- Balzani, V.; Credi, A.; Raymo, F.M.; Stoddart, J.F. Artificial molecular machines. Angew. Chem. Int. Ed. 2000, 39, 3348–3391. [Google Scholar] [CrossRef]
- Diederich, F.; Gomez-Lopez, M. Supramolecular fullerene chemistry. Chem. Soc. Rev. 1999, 28, 263–277. [Google Scholar] [CrossRef]
- Meichsner, E.; Nierengarten, I.; Holler, M.; Chessé, M.; Nierengarten, J.-F. A fullerene-substituted pillar [5]arene for the construction of a photoactive rotaxane. Helv. Chim. Acta 2018, 101, e1800059. [Google Scholar] [CrossRef]
- Steffenhagen, M.; Latus, A.; Trinh, T.M.N.; Nierengarten, I.; Lucas, I.T.; Joiret, S.; Landoulsi, J.; Delavaux-Nicot, B.; Nierengarten, J.-F.; Maisonhaute, E. A rotaxane scaffold bearing multiple redox centers: Synthesis, surface modifications and electrochemical properties. Chem. Eur. J. 2018, 24, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Delavaux-Nicot, B.; Ben Aziza, H.; Nierengarten, I.; Trinh, T.M.N.; Meichsner, E.; Chessé, M.; Holler, M.; Abadi, R.; Maisonhaute, E.; Nierengarten, J.-F. A rotaxane scaffold for the construction of multiporphyrinic light-harvesting devices. Chem. Eur. J. 2018, 24, 133–140. [Google Scholar] [CrossRef] [PubMed]
- de Juan, A.; Pouillon, Y.; Ruiz-Gonzalez, L.; Torres-Pardo, A.; Casado, S.; Martin, N.; Rubio, A.; Perez, E.M. Mechanically Interlocked Single-Wall Carbon Nanotubes. Angew. Chem. Int. Ed. 2014, 53, 5394–5400. [Google Scholar] [CrossRef] [PubMed]
- Barrejon, M.; Mateo-Alonso, A.; Prato, M. Carbon Nanostructures in Rotaxane Architectures. Eur. J. Org. Chem. 2019, 21, 3371–3383. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Diaz, M.V.; Fender, N.S.; Rodriguez-Morgade, M.S.; Gomez-Lopez, M.; Diederich, F.; Echegoyen, L.; Stoddart, J.-F.; Torres, T. A Supramolecular Approach for The Formation of Fullerene–Phthalocyanine Dyads. J. Mat. Chem. 2002, 12, 2095–2099. [Google Scholar] [CrossRef]
- Guldi, D.M.; Ramey, J.; Martinez-Diaz, M.V.; de la Escosura, A.; Torres, T.; Da Ros, T.; Prato, M. Reversible Zinc Phthalocyanine Fullerene Ensembles. Chem. Commun. 2002, 23, 2774–2775. [Google Scholar] [CrossRef]
- Diaz, M.C.; Illescas, M.; Martin, N.; Stoddart, J.-F.; Canales, M.A.; Jimenez-Barbero, J.; Sarova, G.; Guldi, D.M. Supramolecular pseudo-rotaxane type complexes from π-extended TTF dimer crown ether and C60. Tetrahedron 2006, 62, 1998–2002. [Google Scholar] [CrossRef]
- Illescas, B.M.; Santos, J.; Diaz, M.C.; Martin, N.; Atienza, C.M.; Guldi, D.M. Supramolecular Threaded Complexes from Fullerene–Crown Ether and π-Extended TTF Derivatives. Eur. J. Org. Chem. 2007, 30, 5027–5037. [Google Scholar] [CrossRef]
- Watanabe, N.; Kihara, N.; Furusho, Y.; Takata, T.; Araki, Y.; Ito, O. Photoinduced Intrarotaxane Electron Transfer between Zinc Porphyrin and [60]Fullerene in Benzonitrile. Angew. Chem. Int. Ed. 2003, 42, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, T.; Guldi, D.M.; Morales, A.F.; Leigh, D.A.; Prato, M.; Turco, R. Hydrogen Bond-Assembled Fullerene Molecular Shuttle. Org. Lett. 2003, 5, 689–691. [Google Scholar] [CrossRef]
- Mateo-Alonso, A.; Ehli, C.; Rahman, G.M.A.; Guldi, D.M.; Fioravanti, G.; Marcaccio, M.; Paolucci, M.; Prato, M. Tuning Electron Transfer through Translational Motion in Molecular Shuttles. Angew. Chem. Int. Ed. 2007, 46, 3521–3525. [Google Scholar] [CrossRef]
- Mateo-Alonso, A.; Iliopoulos, K.; Couris, S.; Prato, M. Charge Transfer Reactions along a Supramolecular Redox Gradient. J. Am. Chem. Soc. 2008, 130, 14938–14939. [Google Scholar] [CrossRef]
- Saha, S.; Flood, A.H.; Stoddart, J.-F.; Impellizzeri, S.; Silvi, S.; Venturi, M.; Credi, A. A Redox-Driven Multicomponent Molecular Shuttle. J. Am. Chem. Soc. 2007, 129, 12159–12171. [Google Scholar] [CrossRef]
- Ashton, P.R.; Diederich, F.; Gomez-Lopez, M.; Nierengarten, J.-F.; Preece, J.A.; Raymo, F.M.; Stoddart, J.F. Self-Assembly of the First Fullerene-Containing [2]Catenane. Angew. Chem. Int. Ed. 1997, 36, 1448–1451. [Google Scholar] [CrossRef]
- Nakamura, Y.; Minami, S.; Lizuka, K.; Nishimura, J. Preparation of Neutral [60]Fullerene-Based [2]Catenanes and [2]Rotaxanes Bearing an Electron-Deficient Aromatic Diimide Moiety. Angew. Chem. Int. Ed. 2003, 42, 3158–3162. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.T.; Lewis, J.E.M.; Payne, D.T.; D’Souza, F.; Hill, J.P.; Ariga, K.; Yoshikawa, G.; Goldup, S.M. Rotaxanation as a sequestering template to preclude incidental metal insertion in complex oligochromophores. Chem. Commun. 2020, 56, 7447–7450. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, T.; Tanaka, K. Assembly of Multi-Phthalocyanines on a Porphyrin Template by Four fold Rotaxane Formation. Chem. Eur. J. 2006, 22, 12371–12380. [Google Scholar] [CrossRef]
- Barendt, T.A.; Rašović, I.; Lebedeva, M.A.; Farrow, G.A.; Auty, A.; Chekulaev, D.; Sazanovich, I.V.; Weinstein, J.A.; Porfyrakis, K.; Beer, P.D. Anion-Mediated Photophysical Behavior in a C60 Fullerene [3]Rotaxane Shuttle. J. Am. Chem. Soc. 2018, 140, 1924–1936. [Google Scholar] [CrossRef]
- Colasson, B.; Credi, A.; Ventura, B. Photoinduced Electron Transfer Involving a Naphthalimide Chromophore in Switchable and Flexible [2]Rotaxanes. Chem. Eur. J. 2020, 26, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kaur, R.; Wang, B.; Minameyer, M.B.; Gsänger, S.; Meyer, B.; Drewello, T.; Guldi, D.M.; von Delius, M. Concave−Convex π−π Template Approach Enables the Synthesis of [10]Cycloparaphenylene−Fullerene [2]Rotaxanes. J. Am. Chem. Soc. 2018, 140, 13413–13420. [Google Scholar] [CrossRef]
- Browne, C.; Ronson, T.K.; Nitschke, J.R. Palladium-Templated Subcomponent Self-Assembly of Macrocycles, Catenanes, and Rotaxanes. Angew. Chem. Int. Ed. 2014, 53, 10701–10705. [Google Scholar] [CrossRef] [Green Version]
- Alstrum-Acevedo, J.H.; Brennaman, M.K.; Meyer, T.J. Chemical Approaches to Artificial Photosynthesis. 2. Inorg. Chem. 2005, 44, 6802–6827. [Google Scholar] [CrossRef] [Green Version]
- Flamigni, L.; Collin, J.-P.; Sauvage, J.-P. Iridium Terpyridine Complexes as Functional Assembling Units in Arrays for the Conversion of Light Energy. Acc. Chem. Res. 2008, 41, 857–871. [Google Scholar] [CrossRef]
- Hewson, S.W.; Mullen, K.M. Porphyrin-Containing Rotaxane Assemblies. Eur. J. Org. Chem. 2019, 21, 3358–3370. [Google Scholar] [CrossRef]
- Flamigni, L. Photoinduced processes in interlocked structures containing porphyrins. J. Photochem. Photobiol. C Photochem. Rev. 2007, 8, 191–210. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Guldi, D.M.; Schuster, D.I. Fullerenes: Principles and Applications; de la Puente, F.L., Nierengarten, J.F., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; Volume 1, Chapter 10. [Google Scholar]
- Chambron, J.-C.; Heitz, V.; Sauvage, J.-P. Transition metal templated formation of [2]- and [3]-rotaxanes with porphyrins as stoppers. J. Am. Chem. Soc. 1993, 115, 12378–12384. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Harriman, A.; Heitz, V.; Sauvage, J.-P. Ultrafast photoinduced electron transfer between porphyrinic subunits within a bis (porphyrin)-stoppered rotaxane. J. Am. Chem. Soc. 1993, 115, 6109–6114. [Google Scholar] [CrossRef]
- Linke, M.; Chambron, J.-C.; Heitz, V.; Sauvage, J.-P. Electron transfer between mechanically linked porphyrins in a [2]rotaxane. J. Am. Chem. Soc. 1997, 119, 11329–11330. [Google Scholar] [CrossRef]
- Linke, M.; Chambron, J.-C.; Heitz, V.; Sauvage, J.-P.; Semetey, V. Complete rearrangement of a multi-porphyrinic rotaxane by metallation–demetallation of the central coordination site. Chem. Commun. 1998, 23, 2469–2470. [Google Scholar] [CrossRef]
- Solladié, N.; Chambron, J.-C.; Sauvage, J.-P. Porphyrin-stoppered [3]- and [5]-rotaxanes. J. Am. Chem. Soc. 1999, 121, 3684–3692. [Google Scholar] [CrossRef]
- Andersson, M.; Linke, M.; Chambron, J.C.; Davidsson, J.; Heitz, V.; Sauvage, J.-P.; Hammarström, L. Porphyrin-containing [2]-rotaxanes: Metal coordination enhanced superexchange electron transfer between noncovalently linked chromophores. J. Am. Chem. Soc. 2000, 122, 3526–3527. [Google Scholar] [CrossRef]
- Flamigni, L.; Armaroli, N.; Barigelletti, F.; Chambron, J.-C.; Sauvage, J.-P.; Solladié, N. Photoinduced processes in porphyrin-stoppered [3]-rotaxanes. New J. Chem. 1999, 23, 1151–1158. [Google Scholar] [CrossRef]
- Linke, M.; Heitz, V.; Sauvage, J.-P.; Encinas, S.; Barigelletti, F.; Flamigni, L. Multiporphyrinic rotaxanes: Control of intramolecular electron transfer rate by steering the mutual arrangement of the chromophores. J. Am. Chem. Soc. 2000, 122, 11834–11844. [Google Scholar] [CrossRef]
- Andersson, M.; Linke, M.; Chambron, J.C.; Davidsson, J.; Heitz, V.; Hammarström, L.; Sauvage, J.-P. Long-range electron transfer in porphyrin-containing [2]-rotaxanes: Tuning the rate by metal cation coordination. J. Am. Chem. Soc. 2002, 124, 4347–4362. [Google Scholar] [CrossRef]
- Flamigni, L.; Talarico, A.M.; Chambron, J.C.; Heitz, V.; Linke, M.; Fujita, N.; Sauvage, J.-P. Photoinduced Electron Transfer in Multiporphyrinic Interlocked Structures: The Effect of Copper (i) Coordination in the Central Site. Chem. Eur. J. 2004, 10, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Flamigni, L.; Heitz, V.; Sauvage, J.-P. Porphyrin Rotaxanes and Catenanes: Copper (I)-Templated Synthesis and Photoinduced Processes. In Non-Covalent Multi-Porphyrin Assemblies; Alessio, E., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 217–261. [Google Scholar]
- Guldi, D.M.; Prato, M. Excited-State Properties of C60 Fullerene Derivatives. Acc. Chem. Res. 2000, 33, 695–703. [Google Scholar] [CrossRef]
- Guldi, D.M. Fullerene–porphyrin architectures; photosynthetic antenna and reaction center models. Chem. Soc. Rev. 2002, 31, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.F.; Megiatto, J.D., Jr.; Hambourger, M.; Gervaldo, M.; Kodis, G.; Moore, T.A.; Gust, D.; Moore, A.L. Optical and electrochemical properties of hydrogen-bonded phenol-pyrrolidino [60]fullerenes. Photochem. Photobiol. Sci. 2012, 11, 1018–1025. [Google Scholar] [CrossRef]
- Hirsch, A.; Brettreich, M. Fullerenes; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Schuster, D.I.; Megiatto, J.D., Jr. Nanotubes reveal all in solution. Nat. Chem. 2009, 1, 182–183. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Guldi, D.M.; Schuster, D.I. Design, synthesis and photoinduced processes in molecular interlocked photosynthetic [60]fullerene systems. Chem. Soc. Rev. 2020, 49, 8–20. [Google Scholar] [CrossRef]
- Schuster, D.I.; Jarowski, D.A.; Kirschnera, A.N.; Wilson, S.R. Molecular modelling of fullerene–porphyrin dyads. J. Mater. Chem. 2002, 12, 2041–2047. [Google Scholar] [CrossRef]
- Schuster, D.I.; Li, K.; Guldi, D.M.; Palkar, A.; Echegoyen, L.; Stanisky, C.R.; Cross, J.; Niemi, M.; Tkachenko, N.V.; Lemmetyinen, H. Azobenzene-linked porphyrin-fullerene dyads. J. Am. Chem. Soc. 2007, 129, 15973–15982. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking Photosynthetic Solar Energy Transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Molecular mimicry of photosynthetic energy and electron transfer. Acc. Chem. Res. 1993, 26, 198–205. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Diederich, F.; Dietrich-Buchecker, C.O.; Nierengarten, J.-F.; Sauvage, J.-P. A copper (I)-complexed rotaxane with two fullerene stoppers. Chem. Commun. 1995, 7, 781–782. [Google Scholar] [CrossRef]
- Armaroli, N.; Diederich, F.; Dietrich-Buchecker, C.O.; Flamigni, L.; Marconi, G.; Nierengarten, J.-F.; Sauvage, J.-P. A Copper (I)-Complexed Rotaxane with Two Fullerene Stoppers: Synthesis, Electrochemistry, and Photoinduced Processes. Chem. Eur. J. 1998, 4, 406–416. [Google Scholar] [CrossRef]
- Li, K.; Schuster, D.I.; Guldi, D.M.; Herranz, M.A.; Echegoyen, L. Convergent Synthesis and Photophysics of [60]Fullerene/Porphyrin-Based Rotaxanes. J. Am. Chem. Soc. 2004, 126, 3388–3389. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bracher, P.J.; Guldi, D.M.; Herranz, M.A.; Echegoyen, L.; Schuster, D.I. [60]Fullerene-Stoppered Porphyrinorotaxanes: Pronounced Elongation of Charge-Separated-State Lifetimes. J. Am. Chem. Soc. 2004, 126, 9156–9157. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.I.; Li, K.; Guldi, D.M. Porphyrin-fullerene photosynthetic model systems with rotaxane and catenane architectures. C. R. Chim. 2006, 9, 892–908. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Kolb, C.H.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Guldi, D.M.; Schuster, D.I. General method for the synthesis of functionalized macrocycles and catenanes utilizing click chemistry. J. Am. Chem. Soc. 2008, 130, 12872–12873. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Spencer, R.; Schuster, D.I. Efficient one-pot synthesis of rotaxanes bearing electron donors and [60]fullerene. Org. Lett. 2009, 11, 4152–4155. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I. Click methodology for the synthesis of functionalized [3]Catenanes: Toward higher interlocked structures. Chem. A Eur. J. 2009, 15, 5444–5448. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I. Introduction of useful peripheral functional groups on [2]catenanes by combining Cu (I) template synthesis with click chemistry. New J. Chem. 2010, 34, 276–286. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I.; Abwandner, S.; de Miguel, G.; Guldi, D.M. [2]Catenanes decorated with porphyrin and [60]fullerene groups: Design, convergent synthesis, and photoinduced processes. J. Am. Chem. Soc. 2010, 132, 3847–3861. [Google Scholar] [CrossRef] [Green Version]
- Megiatto, J.D., Jr.; Spencer, R.; Schuster, D.I. Optimizing reaction conditions for synthesis of electron donor-[60]fullerene interlocked multiring systems. J. Mater. Chem. 2011, 21, 1544–1550. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I.; de Miguel, G.; Wolfrum, S.; Guldi, D.M. Topological and conformational effects on electron transfer dynamics in porphyrin-[60]fullerene interlocked systems. Chem. Mater. 2012, 24, 2472–2485. [Google Scholar] [CrossRef] [Green Version]
- Kirner, S.V.; Guldi, D.M.; Megiatto, J.D., Jr.; Schuster, D.I. Synthesis and photophysical properties of new catenated electron donor–acceptor materials with magnesium and free base porphyrins as donors and C60 as the acceptor. Nanoscale 2015, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megiatto, J.D., Jr.; Li, K.; Schuster, D.I.; Palkar, A.; Herranz, M.A.; Echegoyen, L.; Abwandner, S.; De Miguel, G.; Guldi, D.M. Convergent synthesis and photoinduced processes in multi-chromophoric rotaxanes. J. Chem. Phys. B 2010, 114, 14408–14419. [Google Scholar] [CrossRef] [Green Version]
- Jakob, M.; Berg, A.; Levanon, H.; Schuster, D.I.; Megiatto, J.D., Jr. Photoexcited state properties of H2-porphyrin/C60-based rotaxanes as studied by time-resolved electron paramagnetic resonance spectroscopy. J. Phys. Chem. A 2011, 115, 5044–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, M.; Berg, A.; Levanon, H.; Schuster, D.I.; Megiatto, J.D., Jr. Photoinduced processes in some mechanically interlocked supramolecules as studied by time-resolved electron paramagnetic resonance. J. Phys. Chem. C 2011, 115, 24555–24563. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I. Alternative demetalation method for Cu (I)-phenanthroline-based catenanes and rotaxanes. Org. Lett. 2011, 13, 1808–1811. [Google Scholar] [CrossRef] [Green Version]
- Megiatto, J.D., Jr.; Méndez-Hernández, D.D.; Tejeda-Ferrari, M.; Teillout, A.-L.; Llansola-Portolés, M.J.; Kodis, G.; Poluektov, O.G.; Rajh, T.; Mujica, V.; Groy, T.L.; et al. A bioinspired redox relay that mimics radical interactions of the tyr-his pairs of photosystem II. Nat. Chem. 2014, 6, 423–428. [Google Scholar] [CrossRef]
- Antoniuk-Pablant, A.; Terazono, Y.; Brennan, B.J.; Sherman, B.D.; Megiatto, J.D., Jr.; Brudvig, G.W.; Moore, A.L.; Moore, T.A.; Gust, D. A new method for the synthesis of β-cyano substituted porphyrins and their use as sensitizers in photoelectrochemical devices. J. Mater. Chem. A 2016, 4, 1–10. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Gervaldo, M.; Moore, T.A.; Moore, A.L.; Gust, D. Mimicking the electron transfer chain in photosystem ii with a molecular triad thermodynamically capable of water oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15578–15583. [Google Scholar] [CrossRef] [Green Version]
- Ravensbergen, J.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Megiatto, J.D., Jr.; Méndez-Hernández, D.D.; Frese, R.N.; Van Grondelle, R.; Moore, T.A.; Moore, A.L.; et al. Spectroscopic analysis of a biomimetic model of Tyr function in PSII. J. Phys. Chem. B 2015, 119, 12156–12163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Swierk, J.R.; Megiatto, J.D., Jr.; Sherman, B.; Youngblood, W.J.; Qin, D.; Lentz, D.M.; Moore, A.L.; Moore, T.A.; Gust, D.; et al. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator. Proc. Natl. Acad. Sci. USA 2012, 109, 15612–15616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megiatto, J.D., Jr.; Patterson, D.; Sherman, B.D.; Moore, T.A.; Gust, D.; Moore, A.L. Intramolecular hydrogen bonding as a synthetic tool to induce chemical selectivity in acid catalyzed porphyrin synthesis. Chem. Commun. 2012, 48, 4558–4560. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.A.; Rigolin, V.H.; Braga, C.B.; Ornelas, C.; Megiatto, J.D., Jr. Methodology for functionalization of water oxidation catalyst IrOx nanoparticles with hydrophobic and multi-functionalized chromophores. Chem. Commun. 2021, 57, 7398–7401. [Google Scholar] [CrossRef] [PubMed]
- Kirner, S.V.; Henkel, C.; Guldi, D.M.; Megiatto, J.D., Jr.; Schuster, D.I. Multistep energy and electron transfer processes in novel rotaxane donor-acceptor hybrids generating microsecond-lived charge separated states. Chem. Sci. 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chambron, J.-C.; Collin, J.P.; Heitz, V.; Jouvenot, D.; Kern, J.-M.; Mobian, P.; Pomeranc, D.; Sauvage, J.-P. Rotaxanes and Catenanes Built Around Octahedral Transition Metals. Eur. J. Org. Chem. 2004, 8, 1627–1638. [Google Scholar] [CrossRef]
- Alcantara, A.F.P.; Fontana, L.A.; Rigolin, V.H.; Andrade, Y.S.F.; Ribeiro, M.A.; Barros, W.P.; Ornelas, C.; Megiatto, J.D., Jr. Olefin cyclopropanation via radical carbene transfer reaction promoted by Co (II) porphyrinates for the active-metal-template synthesis of [2]rotaxanes. Angew. Chem. Int. Ed. 2018, 57, 8979–8983. [Google Scholar] [CrossRef]
- Alcantara, A.F.P.; Fontana, L.A.; Almeida, M.P.; Rigolin, V.H.; Ribeiro, M.A.; Barros, W.P.; Megiatto, J.D., Jr. Control over the Redox Cooperative Mechanism of Radical Carbene Transfer Reactions for the Efficient Active-Metal-Template Synthesis of [2]Rotaxanes. Chem. Eur. J. 2020, 26, 7808–7822. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.A.; Almeida, M.P.; Alcantara, A.F.P.; Rigolin, V.H.; Ribeiro, M.A.; Barros, W.P.; Megiatto, J.D. Ru(II)Porphyrinate-based molecular nanoreactor for carbene insertion reactions and quantitative formation of rotaxanes by active-metal-template syntheses. Nat. Commun. 2020, 11, 6370. [Google Scholar] [CrossRef]
- Fontana, L.A.; Alcantara, A.F.P.; Rigolin, V.H.; Megiatto, J.D., Jr. Semi-Rigid Ru (II) Porphyrinate-Based Macrocyclic Receptor as Endotopic Catalyst for Carbene Transfer Reactions. ECS J. Solid State Sci. Technol. 2020, 9, 061023. [Google Scholar] [CrossRef]
- van den Boomen, O.I.; Coumans, R.G.E.; Akeroyd, N.; Peters, T.P.J.; Schlebos, P.P.J.; Smits, J.; de Gelder, R.; Elemans, J.A.A.W.; Nolte, R.J.M.; Rowan, A.E. Carbenoid transfer reactions catalyzed by a ruthenium porphyrin macrocycle. Tetrahedron 2017, 73, 5029–5037. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; Kahlfuss, C.; Ogawa, A.; Matsumoto, T.; Wytko, J.A.; Oohora, K.; Hayashi, T.; Weiss, J. CuAAC in a Distal Pocket: Metal Active-Template Synthesis of Strapped-Porphyrin [2]Rotaxanes. Chem. Eur. J. 2017, 23, 13579–13582. [Google Scholar] [CrossRef]
- Lewis, J.E.M.; Modicom, F.; Goldup, S.M. Efficient Multi-Component Active Template Synthesis of Catenanes. J. Am. Chem. Soc. 2018, 140, 4787–4791. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.; Goldup, S.M. The Active Template Approach to Interlocked Molecules: Principles, Progress and Applications. Nat. Rev. Chem. 2017, 1, 0061. [Google Scholar] [CrossRef]
- Lewis, J.E.M.; Winn, J.; Cera, L.; Goldup, S.M. Iterative Synthesis of Oligo[n]Rotaxanes in Excellent Yield. J. Am. Chem. Soc. 2016, 138, 16329–16336. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Jake, A.; Ball, A.T.; Galli, M.; Kukwikila, M.; Denis, M.; Singleton, D.G.; Tavassoli, A.; Goldup, S.M. AT-CuAAC Synthesis of Mechanically Interlocked Oligonucleotides. J. Am. Chem. Soc. 2020, 142, 5985–5990. [Google Scholar] [CrossRef] [PubMed]
- Danon, J.J.; Leigh, D.A.; McGonigal, P.R.; Ward, J.W.; Wu, J. Triply-threaded [4]rotaxanes. J. Am. Chem. Soc. 2016, 138, 12643–12674. [Google Scholar] [CrossRef] [Green Version]
- Thordarson, P.; Bijsterveld, E.J.A.; Rowan, A.E.; Nolte, R.J.M. Epoxidation of polybutadiene by a topologically linked catalyst. Nature 2003, 424, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Echavarren, J.; Gall, M.A.Y.; Haertsch, A.; Leigh, D.A.; Spence, J.T.J.; Tetlow, D.J.; Tian, C. Sequence-selective decapeptide synthesis by the parallel operation of two artificial molecular machines. J. Am. Chem. Soc. 2021, 143, 5158–5165. [Google Scholar] [CrossRef]
- Amano, S.; Fielden, S.D.P.; Leigh, D.A. A catalysis-driven artificial molecular pump. Nature 2021, 594, 529–534. [Google Scholar] [CrossRef]
- McTernan, C.; De Bo, G.; Leigh, D.A. A track-based molecular synthesizer that builds a single-sequence oligomer through iterative carbon-carbon bond formation. Chem 2020, 6, 2964–2973. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Fielden, S.D.P.; Karaca, U.; Leigh, D.A.; McTernan, C.T.; Tetlow, D.J.; Wilson, M.R. Rotary and linear molecular motors driven by pulses of a chemical fuel. Science 2017, 358, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, M.; Ogawa, A.; Bechtold, M.; Vonesch, M.; Wytko, J.A.; Oohora, K.; Campidelli, S.; Hayashi, T.; Guldi, D.M.; Weiss, J. Light triggers molecular shuttling in rotaxanes: Control over proximity and charge recombination. Chem. Sci. 2019, 10, 3846–3853. [Google Scholar] [CrossRef] [Green Version]
- Kohn, D.R.; Movsisyan, L.D.; Thompson, A.L.; Anderson, H.L. Porphyrin–Polyyne [3]- and [5]Rotaxanes. Org. Lett. 2017, 19, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Movsisyan, L.D.; Franz, M.; Hampel, F.; Thompson, A.L.; Tykwinski, R.R.; Anderson, H.L. Polyyne Rotaxanes: Stabilization by Encapsulation. J. Am. Chem. Soc. 2016, 138, 1366–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruchyathamkorn, J.; Kendrick, W.J.; Frawley, A.T.; Mattioni, A.; Caycedo-Soler, F.; Huelga, S.F.; Plenio, M.B.; Anderson, H.L. A cyanine dye rotaxane porphyrin nanoring complex as a model light harvesting system. Angew. Chem. Int. Ed. 2020, 59, 16455–16458. [Google Scholar] [CrossRef]
- Movsisyan, L.D.; Kondratuk, D.V.; Franz, M.; Thompson, A.L.; Tykwinski, R.R.; Anderson, A.L. Synthesis of Polyyne Rotaxanes. Org. Lett. 2012, 14, 3424–3426. [Google Scholar] [CrossRef]
- Movsisyan, L.D.; Peeks, M.D.; Greetham, G.M.; Towrie, M.; Thompson, A.L.; Parker, A.W.; Anderson, H.L. Photophysics of Threaded sp-Carbon Chains: The Polyyne is a Sink for Singlet and Triplet Excitation. J. Am. Chem. Soc. 2014, 136, 17996–18008. [Google Scholar] [CrossRef] [PubMed]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigolin, V.H.; Fontana, L.A.; Megiatto, J.D., Junior. A Brief History of Photoactive Interlocked Systems Assembled by Transition Metal Template Synthesis. Photochem 2021, 1, 411-433. https://doi.org/10.3390/photochem1030025
Rigolin VH, Fontana LA, Megiatto JD Junior. A Brief History of Photoactive Interlocked Systems Assembled by Transition Metal Template Synthesis. Photochem. 2021; 1(3):411-433. https://doi.org/10.3390/photochem1030025
Chicago/Turabian StyleRigolin, Vitor H., Liniquer A. Fontana, and Jackson D. Megiatto, Junior. 2021. "A Brief History of Photoactive Interlocked Systems Assembled by Transition Metal Template Synthesis" Photochem 1, no. 3: 411-433. https://doi.org/10.3390/photochem1030025
APA StyleRigolin, V. H., Fontana, L. A., & Megiatto, J. D., Junior. (2021). A Brief History of Photoactive Interlocked Systems Assembled by Transition Metal Template Synthesis. Photochem, 1(3), 411-433. https://doi.org/10.3390/photochem1030025