
Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes


Abstract
:1. Introduction
2. Computational Methods
3. Results
3.1. Geometry Optimization
3.2. Modelling Spectroscopic Properties
3.3. Fluorescence
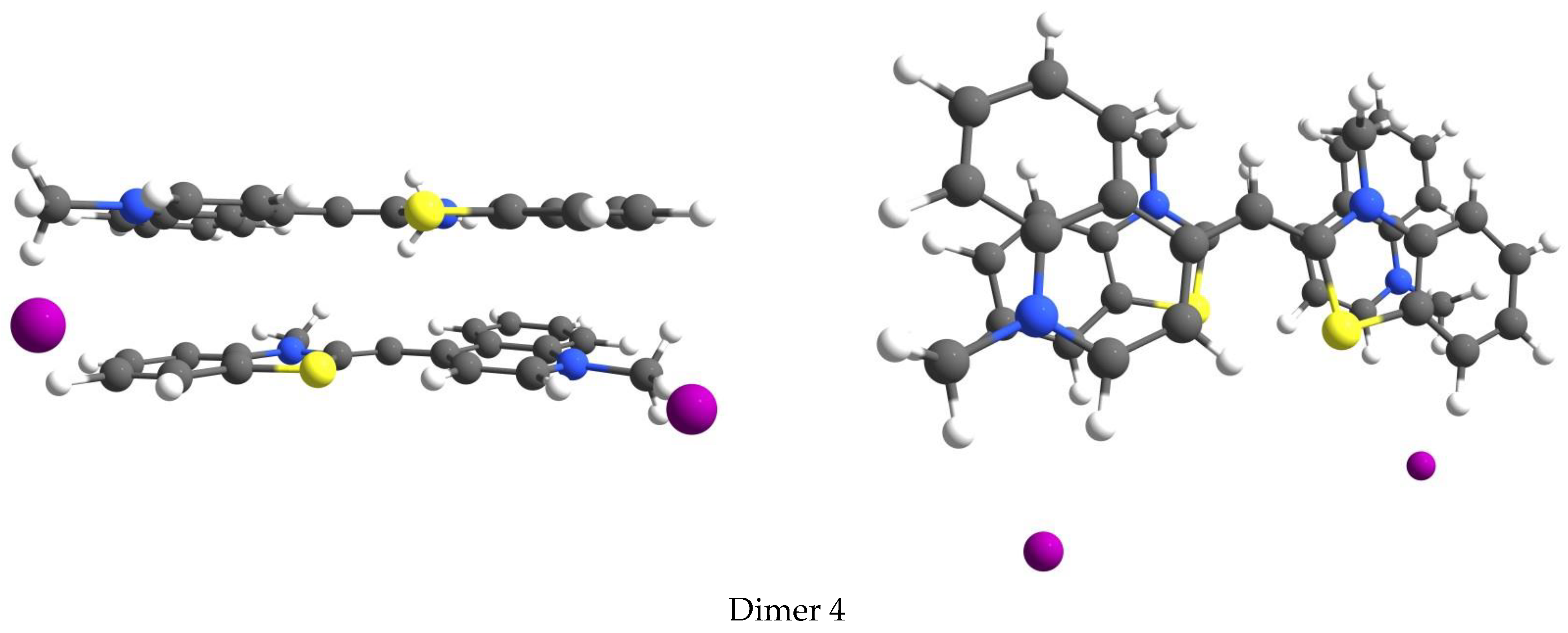
3.4. Aggregation of the Dyes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pindur, U.; Jansen, M.; Lemster, T. Advances in DNA-Ligands with Groove Binding, Intercalating and/or Alkylating Activity: Chemistry, DNA-Binding and Biology. Curr. Med. Chem. 2005, 12, 2805–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rask-Andersen, M.; Almén, M.S.; Schiöth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Saarnio, V.K.; Salorinne, K.; Ruokolainen, V.P.; Nilsson, J.R.; Tero, T.-R.; Oikarinen, S.; Wilhelmsson, L.M.; Lahtinen, T.M.; Marjomäki, V.S. Development of functionalized SYBR green II related cyanine dyes for viral RNA detection. Dye. Pigment. 2020, 177, 108282. [Google Scholar] [CrossRef]
- Johnson, I.; Spence, M.T.Z. The Molecular Probes Handbook—A Guide to Fluorescent Probes and Labeling Technologies, 11th ed.; Life Technologies Corporation: Carlsbad, CA, USA, 2010. [Google Scholar]
- Spielmann, H.P.; Wemmer, D.E.; Jacobsen, J.P. Solution Structure of a DNA Complex with the Fluorescent Bis-Intercalator TOTO Determined by NMR Spectroscopy. Biochemistry 1995, 34, 8542–8553. [Google Scholar] [CrossRef] [PubMed]
- Yarmoluk, S.M.; Lukashov, S.S.; Ogul’Chansky, T.Y.; Losytskyy, M.Y.; Kornyushyna, O.S. Interaction of cyanine dyes with nucleic acids. XXI. Arguments for half-intercalation model of interaction. Biopolymers 2001, 62, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Zhytniakivska, O.; Girych, M.; Trusova, V.; Gorbenko, G.; Vasilev, A.; Kandinska, M.; Kurutos, A.; Baluschev, S.B. Spectroscopic and molecular docking studies of the interactions of monomeric unsymmetrical polycationic fluorochromes with DNA and RNA. Dye. Pigment. 2020, 180, 108446. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Moradi, R.; Lashgari, N.; Kruger, H.G. Cyanine Dyes. In Metal-Free Synthetic Organic Dyes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 127–152. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine dye-DNA interactions: Intercalation, groove binding, and aggregation. In DNA Binders and Related Subjects; Waring, M.J., Chaires, J.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 253, pp. 55–76. ISBN 3540228357. [Google Scholar]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2012. [Google Scholar] [CrossRef]
- Silva, G.L.; Ediz, V.; Yaron, D.; Armitage, B.A. Experimental and Computational Investigation of Unsymmetrical Cyanine Dyes: Understanding Torsionally Responsive Fluorogenic Dyes. J. Am. Chem. Soc. 2007, 129, 5710–5718. [Google Scholar] [CrossRef] [Green Version]
- Rastede, E.E.; Tanha, M.; Yaron, D.; Watkins, S.C.; Waggoner, A.S.; Armitage, B.A. Spectral fine tuning of cyanine dyes: Electron donor–acceptor substituted analogues of thiazole orange. Photochem. Photobiol. Sci. 2015, 14, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Biancardi, A.; Biver, T.; Marini, A.; Mennucci, B.; Secco, F. Thiazole orange (TO) as a light-switch probe: A combined quantum-mechanical and spectroscopic study. Phys. Chem. Chem. Phys. 2011, 13, 12595–12602. [Google Scholar] [CrossRef]
- Jacquemin, D.; Zhao, Y.; Valero, R.; Adamo, C.; Ciofini, I.; Truhlar, D.G. Verdict: Time-Dependent Density Functional Theory “Not Guilty” of Large Errors for Cyanines. J. Chem. Theory Comput. 2012, 8, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Vus, K.; Girych, M.; Trusova, V.; Gorbenko, G.; Kurutos, A.; Vasilev, A.; Gadjev, N.; Deligeorgiev, T. Cyanine dyes derived inhibition of insulin fibrillization. J. Mol. Liq. 2019, 276, 541–552. [Google Scholar] [CrossRef]
- Gorbenko, G.; Trusova, V.; Kirilova, E.; Kirilov, G.; Kalnina, I.; Vasilev, A.; Kaloyanova, S.; Deligeorgiev, T. New fluorescent probes for detection and characterization of amyloid fibrils. Chem. Phys. Lett. 2010, 495, 275–279. [Google Scholar] [CrossRef]
- Vasilev, A.; Deligeorgiev, T.; Kaloyanova, S.; Stoyanov, S.; Maximova, V.; Vaquero, J.J.; Alvarez-Builla, J. Synthesis of novel tetracationic asymmetric monomeric monomethine cyanine dyes—Highly fluorescent dsDNA probes. Color. Technol. 2011, 127, 69–74. [Google Scholar] [CrossRef]
- Kandinska, M.; Cheshmedzhieva, D.; Kostadinov, A.; Rusinov, K.; Rangelov, M.; Todorova, N.; Ilieva, S.; Ivanov, D.; Videva, V.; Lozanov, V.; et al. Tricationic asymmetric monomeric monomethine cyanine dyes with chlorine and trifluoromethyl functionality—Fluorogenic nucleic acids probes. J. Mol. Liq. 2021, 342, 117501. [Google Scholar] [CrossRef]
- Sato, Y. Design of Fluorescent Peptide Nucleic Acid Probes Carrying Cyanine Dyes for Targeting Double-Stranded RNAs for Analytical Applications. Bull. Chem. Soc. Jpn. 2020, 93, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Martineau, C.; Whyte, L.G.; Greer, C.W. Development of a SYBR safe™ technique for the sensitive detection of DNA in cesium chloride density gradients for stable isotope probing assays. J. Microbiol. Methods 2008, 73, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.F.; Kelso, S.; Skevington, J.H. Band-cutting no more: A method for the isolation and purification of target PCR bands from multiplex PCR products using new technology. Mol. Phylogenetics Evol. 2010, 56, 1126–1128. [Google Scholar] [CrossRef]
- Clauß, M.; Springorum, A.C.; Hartung, J. Comparison of Different Fluorescence and Non-Fluorescence Staining Techniques for Rapid Detection of Airborne Micro-Organisms Collected on Room Temperature Vulcanizing (RTV) Silicones from Generated Aerosols and from Ambient Air. Aerosol Sci. Technol. 2012, 46, 818–827. [Google Scholar] [CrossRef]
- Pérez-Cordero, J.-J.; Sánchez-Suárez, J.; Delgado, G. Use of a fluorescent stain for evaluating in vitro infection with Leishmania panamensis. Exp. Parasitol. 2011, 129, 31–35. [Google Scholar] [CrossRef]
- Send, R.; Valsson, O.; Filippi, C. Electronic Excitations of Simple Cyanine Dyes: Reconciling Density Functional and Wave Function Methods. J. Chem. Theory Comput. 2011, 7, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Charaf-Eddin, A.; Le Guennic, B.; Jacquemin, D. Excited-states of BODIPY–cyanines: Ultimate TD-DFT challenges? RSC Adv. 2014, 4, 49449–49456. [Google Scholar] [CrossRef]
- Grimme, S.; Neese, F. Double-hybrid density functional theory for excited electronic states of molecules. J. Chem. Phys. 2007, 127, 154116. [Google Scholar] [CrossRef] [PubMed]
- Zhekova, H.; Krykunov, M.; Autschbach, J.; Ziegler, T. Applications of Time Dependent and Time Independent Density Functional Theory to the First π to π* Transition in Cyanine Dyes. J. Chem. Theory Comput. 2014, 10, 3299–3307. [Google Scholar] [CrossRef]
- Le Guennic, B.; Jacquemin, D. Taking Up the Cyanine Challenge with Quantum Tools. Acc. Chem. Res. 2015, 48, 530–537. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Cheshmedzhieva, D.; Ivanova, P.; Stoyanov, S.; Tasheva, D.; Dimitrova, M.; Ivanov, I.; Ilieva, S. Experimental and theoretical study on the absorption and fluorescence properties of substituted aryl hydrazones of 1,8-naphthalimide. Phys. Chem. Chem. Phys. 2011, 13, 18530–18538. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpète, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C.; Valero, R.; Zhao, Y.; Truhlar, D. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. J. Chem. Theory Comput. 2010, 6, 2071–2085. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scalmani, G.; Frisch, M.J.; Kobayashi, R.; Adamo, C. Assessment of the efficiency of long-range corrected functionals for some properties of large compounds. J. Chem. Phys. 2007, 126, 144105. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Fülscher, M.; Malmqvist, P.-Å.; Merchán, M.; Serrano-Andrés, L. Theoretical Studies of the Electronic Spectra of Organic Molecules. In Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy; Springer: Dordrecht, The Netherlands, 1995; pp. 357–438. [Google Scholar]
- Truhlar, D.G. Valence bond theory for chemical dynamics. J. Comput. Chem. 2007, 28, 73–86. [Google Scholar] [CrossRef]
- Champagne, B.; Guillaume, M.; Zutterman, F. TDDFT investigation of the optical properties of cyanine dyes. Chem. Phys. Lett. 2006, 425, 105–109. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision 16.A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Labanowski, J.K.; Andzelm, J.W. (Eds.) Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991; ISBN 978-1-4612-7809-2. [Google Scholar]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; De Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, A.M.; Schreckenbach, G. Computational study of the ground state properties of iodine and polyiodide ions. Theor. Chem. Acta 2009, 122, 119–125. [Google Scholar] [CrossRef]
- Lin, R.Y.-Y.; Chuang, T.-M.; Wu, F.-L.; Chen, P.-Y.; Chu, T.-C.; Ni, J.-S.; Fan, M.-S.; Lo, Y.-H.; Ho, K.-C.; Lin, J.T. Anthracene/Phenothiazine π-Conjugated Sensitizers for Dye-Sensitized Solar Cells using Redox Mediator in Organic and Water-based Solvents. ChemSusChem 2015, 8, 105–113. [Google Scholar] [CrossRef]
- Sohrabi, M.; Amirnasr, M.; Farrokhpour, H.; Meghdadi, S. A single chemosensor with combined ionophore/fluorophore moieties acting as a fluorescent “Off-On” Zn2+ sensor and a colorimetric sensor for Cu2+: Experimental, logic gate behavior and TD-DFT calculations. Sens. Actuators B Chem. 2017, 250, 647–658. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242–250. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Kaloyanova, S.; Ivanova, I.; Tchorbanov, A.; Dimitrova, P.; Deligeorgiev, T. Synthesis of chloro-substituted analogs of Thiazole orange—Fluorophores for flow cytometric analyses. J. Photochem. Photobiol. B Biol. 2011, 103, 215–221. [Google Scholar] [CrossRef]
- Vasilev, A.A.; Kandinska, M.I.; Stoyanov, S.S.; Yordanova, S.B.; Sucunza, D.; Vaquero, J.J.; Castaño, O.D.; Baluschev, S.; Angelova, S.E. Halogen-containing thiazole orange analogues—New fluorogenic DNA stains. Beilstein J. Org. Chem. 2017, 13, 2902–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evenson, W.E.; Boden, L.M.; Muzikar, K.A.; O’Leary, D.J. 1H and 13C NMR Assignments for the Cyanine Dyes SYBR Safe and Thiazole Orange. J. Org. Chem. 2012, 77, 10967–10971. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Casida, M.E. Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods; Recent Advances in Computational Chemistry; World Scientific: Singapore, 1995; pp. 155–192. [Google Scholar] [CrossRef]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys. 2011, 13, 16987–16998. [Google Scholar] [CrossRef]
- Toulouse, J.; Colonna, F.; Savin, A. Long-range–short-range separation of the electron-electron interaction in density-functional theory. Phys. Rev. A 2004, 70, 062505. [Google Scholar] [CrossRef] [Green Version]
- Seminario, J.M. (Ed.) Recent Developments and Applications of Modern Density Functional Theory; Elsevier: Amsterdam, The Netherlands, 1996; pp. 327–354. ISBN 9780444824042. [Google Scholar]
- Oberhofer, K.E.; Musheghyan, M.; Wegscheider, S.; Wörle, M.; Iglev, E.D.; Nikolova, R.D.; Kienberger, R.; Pekov, P.S.; Iglev, H. Individual control of singlet lifetime and triplet yield in halogen-substituted coumarin derivatives. RSC Adv. 2020, 10, 27096–27102. [Google Scholar] [CrossRef]
- Hunt, P.A.; Robb, M.A. Systematic Control of Photochemistry: The Dynamics of Photoisomerization of a Model Cyanine Dye. J. Am. Chem. Soc. 2005, 127, 5720–5726. [Google Scholar] [CrossRef]
- Yang, D.; Liu, Y.; Shi, D.; Sun, J. Theoretical study on the excited-state photoinduced electron transfer facilitated by hydrogen bonding strengthening in the C337–AN/MAN complexes. Comput. Theor. Chem. 2012, 984, 76–84. [Google Scholar] [CrossRef]
- Pedone, A. Role of Solvent on Charge Transfer in 7-Aminocoumarin Dyes: New Hints from TD-CAM-B3LYP and State Specific PCM Calculations. J. Chem. Theory Comput. 2013, 9, 4087–4096. [Google Scholar] [CrossRef]
- Karunakaran, V.; Lustres, J.L.P.; Zhao, L.; Ernsting, N.P.; Seitz, O. Large Dynamic Stokes Shift of DNA Intercalation Dye Thiazole Orange has Contribution from a High-Frequency Mode. J. Am. Chem. Soc. 2006, 128, 2954–2962. [Google Scholar] [CrossRef]
- Sanchez-Galvez, A.; Hunt, P.; Robb, M.A.; Olivucci, M.; Vreven, A.T.; Schlegel§, H.B. Ultrafast Radiationless Deactivation of Organic Dyes: Evidence for a Two-State Two-Mode Pathway in Polymethine Cyanines. J. Am. Chem. Soc. 2000, 122, 2911–2924. [Google Scholar] [CrossRef]
- Retting, W. Charge Separation in Excited States of Decoupled Systems—TICT Compounds and Implications Regarding the Development of New Laser Dyes and the Primary Process of Vision and Photosynthesis. Angew. Chem. Int. Ed. Engl. 1986, 25, 971–988. [Google Scholar] [CrossRef]
- West, W.; Pearce, S. The Dimeric State of Cyanine Dyes. J. Phys. Chem. 1965, 69, 1894–1903. [Google Scholar] [CrossRef]
- Biver, T.; Boggioni, A.; Secco, F.; Turriani, E.; Venturini, M.; Yarmoluk, S. Influence of cyanine dye structure on self-aggregation and interaction with nucleic acids: A kinetic approach to TO and BO binding. Arch. Biochem. Biophys. 2007, 465, 90–100. [Google Scholar] [CrossRef]
- Ogul’Chansky, T.; Losytskyy, M.; Kovalska, V.; Yashchuk, V.; Yarmoluk, S. Interactions of cyanine dyes with nucleic acids. XXIV. Aggregation of monomethine cyanine dyes in presence of DNA and its manifestation in absorption and fluorescence spectra. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2001, 57, 1525–1532. [Google Scholar] [CrossRef]
- Nygren, J.; Svanvik, N.; Kubista, M. The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers 1998, 46, 39–51. [Google Scholar] [CrossRef]
- Mooi, S.M.; Heyne, B. Size Does Matter: How To Control Organization of Organic Dyes in Aqueous Environment Using Specific Ion Effects. Langmuir 2012, 28, 16524–16530. [Google Scholar] [CrossRef]
- Mooi, S.M.; Keller, S.N.; Heyne, B. Forcing Aggregation of Cyanine Dyes with Salts: A Fine Line between Dimers and Higher Ordered Aggregates. Langmuir 2014, 30, 9654–9662. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Isomer | τ1 (NC2C3H) | τ2 (HC3C4C5) | τ (C2C3C4C6) | ∆G (kcal/mol) | λabs (nm) | f |
| cis | −14.8 | 157.7 | 145.6 | 5.4 | 476.8 | 0.6706 |
| trans | 6.4 | 10.0 | 12.7 | 0.0 | 447.2 | 0.9598 |
| trans 2 | 6.5 | 9.1 | 11.8 | 0.8 | 448.8 | 0.9000 |
| trans 3 | −6.5 | −10.6 | −13.2 | 1.5 | 447.2 | 0.9504 |
| Basis Set | λabs [nm] PCM | λabs [nm] SMD |
|---|---|---|
| 6-31G(d,p) | 445 | 445 |
| 6-31+G(d,p) | 454 | 453 |
| 6-31++G(d,p) | 454 | 453 |
| 6-311G(d,p) | 451 | 450 |
| 6-311+G(d,p) | 452 | 455 |
| 6-311+G(2d,p) | 458 | 457 |
| Experiment λabs (nm) | 502 a | |
| Dye | B3LYP | PBE0 | M062X | BH&HLYP | CAM B3LYP | M06 | M06L | HFS | HFB | B97D |
|---|---|---|---|---|---|---|---|---|---|---|
| TO | 0.24 | 0.30 | 0.39 | 0.52 | 0.42 | 0.27 | 0.20 | 0.01 | 0.02 | 0.06 |
| 1b | 0.25 | 0.31 | 0.40 | 0.54 | 0.44 | 0.29 | 0.21 | 0.01 | 0.04 | 0.06 |
| B9 | 0.23 | 0.29 | 0.39 | 0.52 | 0.43 | 0.26 | 0.16 | −0.03 | −0.01 | 0.03 |
| B11 | 0.23 | 0.29 | 0.38 | 0.52 | 0.42 | 0.26 | 0.16 | −0.03 | −0.01 | 0.03 |
| B13 | 0.22 | 0.28 | 0.38 | 0.52 | 0.42 | 0.25 | 0.16 | −0.06 | −0.03 | 0.01 |
| 6b | 0.11 | 0.17 | 0.33 | 0.41 | 0.35 | 0.15 | 0.07 | −0.11 | −0.09 | −0.07 |
| 7Cl−TO | 0.24 | 0.31 | 0.39 | 0.52 | 0.42 | 0.28 | 0.19 | 0.00 | 0.02 | 0.05 |
| sof-5 | 0.19 | 0.26 | 0.39 | 0.51 | 0.43 | 0.23 | 0.04 | −0.17 | −0.13 | −0.11 |
| MAD a | 0.21 | 0.28 | 0.38 | 0.51 | 0.42 | 0.25 | 0.15 | −0.05 | −0.02 | 0.01 |
| TO Dimer | ΔG (kcal/mol) | λabs (nm) a |
|---|---|---|
| Dimer 1 | −1.4 | 415 |
| Dimer 2 | −6.1 | 423 |
| Dimer 3 | −5.9 | 417 |
| Dimer 4 | −4.6 | 426 |
| Method/Basis Set | Monomer | Dimer |
|---|---|---|
| HFS/6-311+G(2d,p) | 500 | 484 |
| HFB/6-311+G(2d,p) | 497 | 477 |
| PBE0/6-311+G(2d,p) | 447 | 423 |
| experiment | 501 | 471 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, S.; Kandinska, M.; Vasilev, A.; Cheshmedzhieva, D. Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes. Photochem 2022, 2, 202-216. https://doi.org/10.3390/photochem2010015
Ilieva S, Kandinska M, Vasilev A, Cheshmedzhieva D. Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes. Photochem. 2022; 2(1):202-216. https://doi.org/10.3390/photochem2010015
Chicago/Turabian StyleIlieva, Sonia, Meglena Kandinska, Aleksey Vasilev, and Diana Cheshmedzhieva. 2022. "Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes" Photochem 2, no. 1: 202-216. https://doi.org/10.3390/photochem2010015
APA StyleIlieva, S., Kandinska, M., Vasilev, A., & Cheshmedzhieva, D. (2022). Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes. Photochem, 2(1), 202-216. https://doi.org/10.3390/photochem2010015