
Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution



Abstract
:1. Introduction
2. Materials and Methods
2.1. Starting Materials and Preparation of Samples
2.2. Instrumentation
2.3. Computational Details
3. Results and Discussion
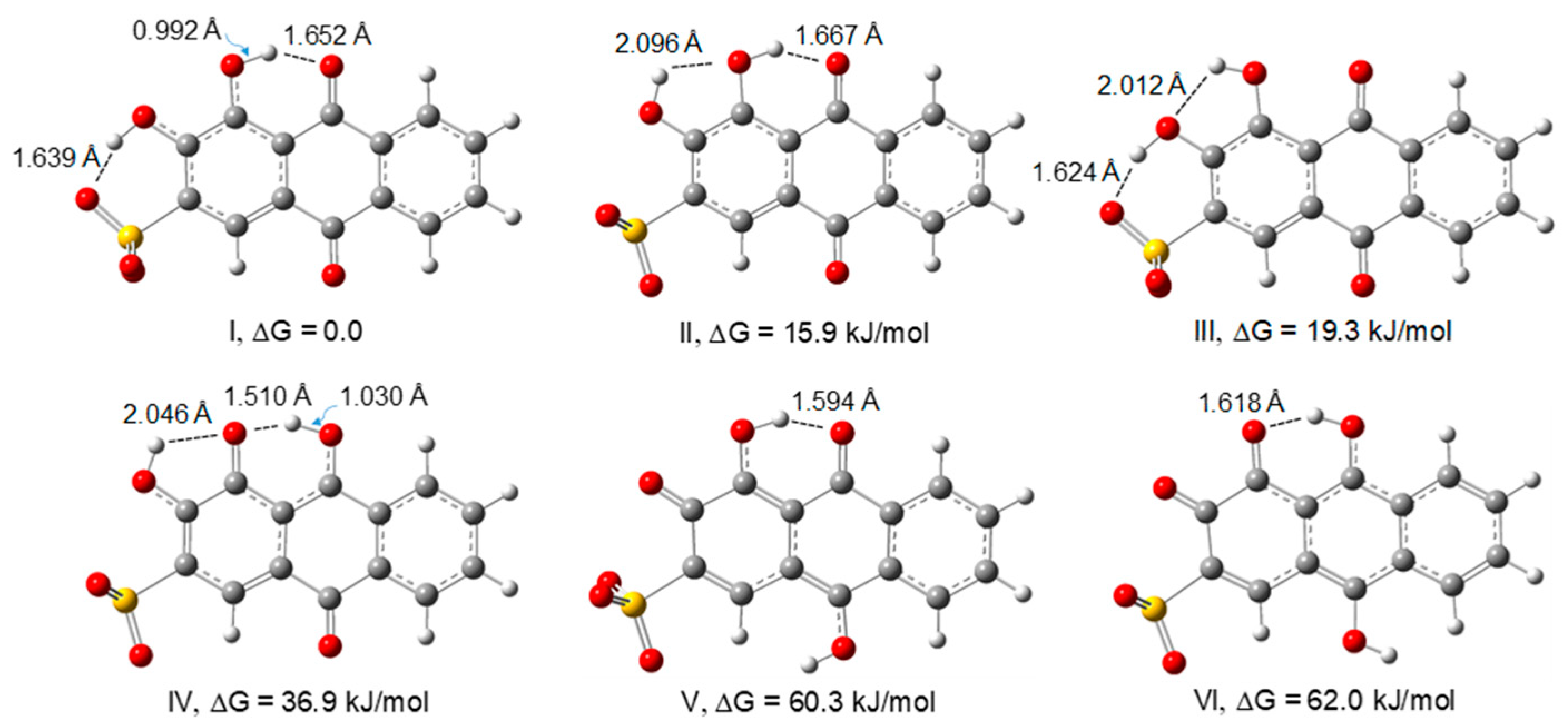
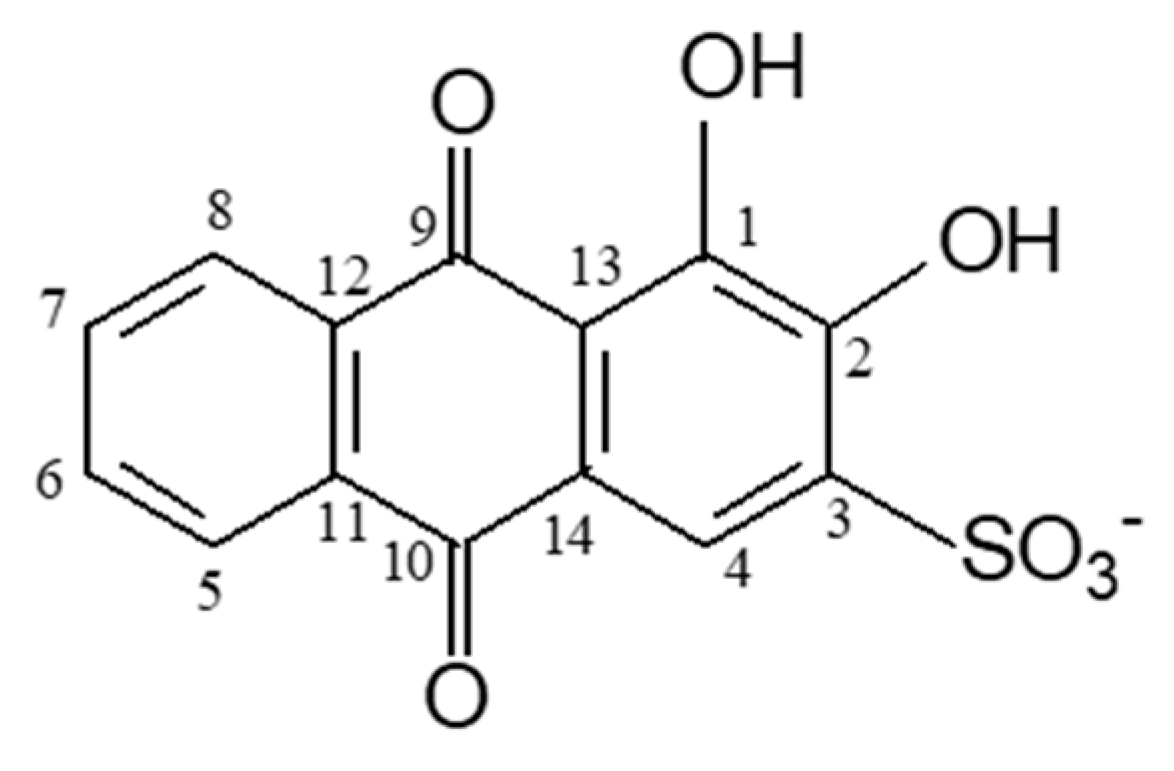
3.1. Structure and Energetics of Alizarin Red S
3.2. Complexation between Ga(III) and 1,2-Dihydroxy-9,10-anthraquinone-3-sulfonate (ARS)
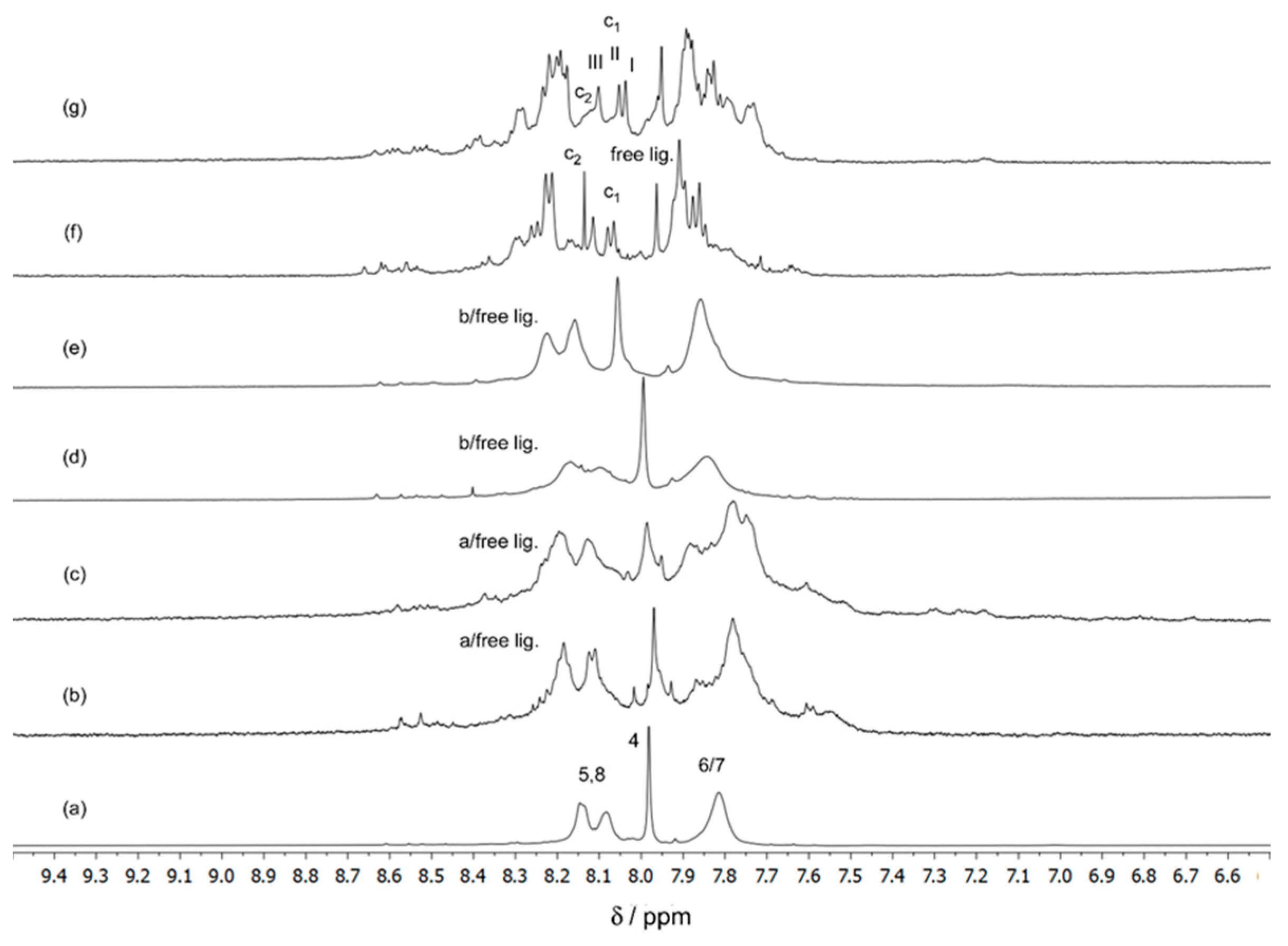
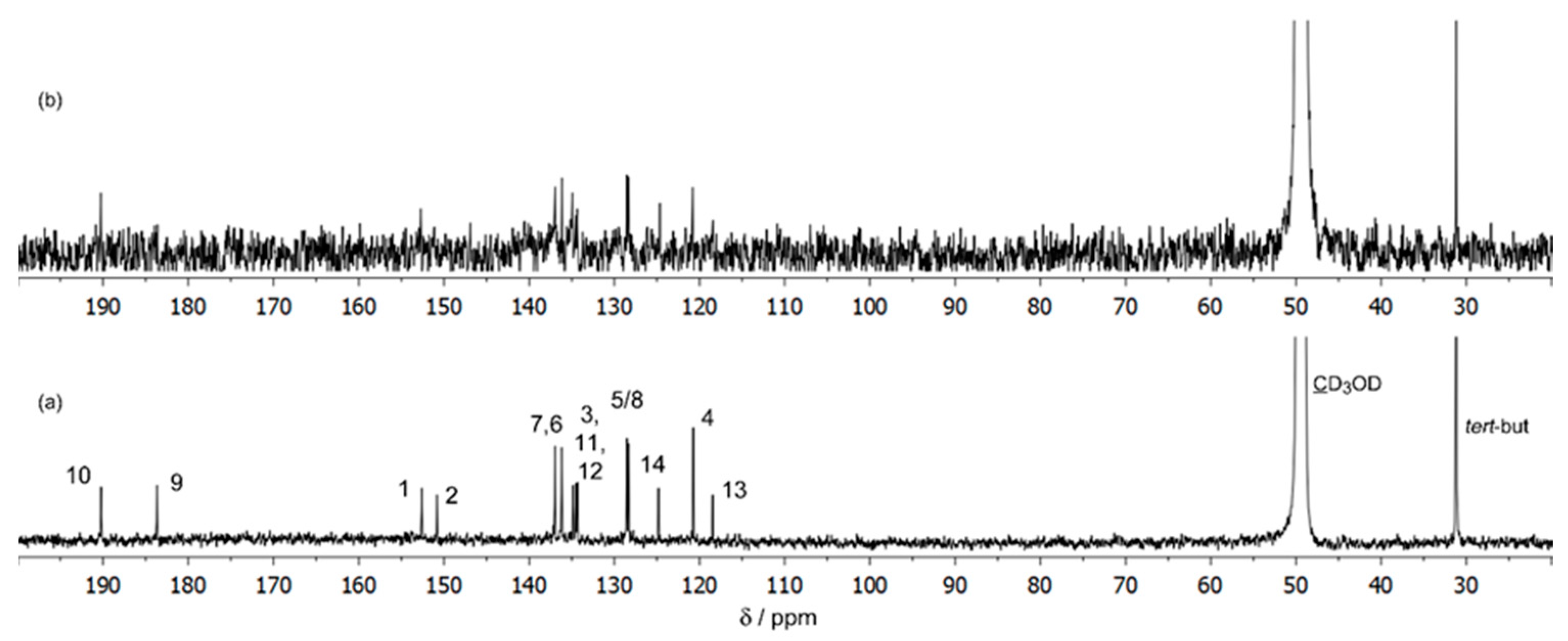
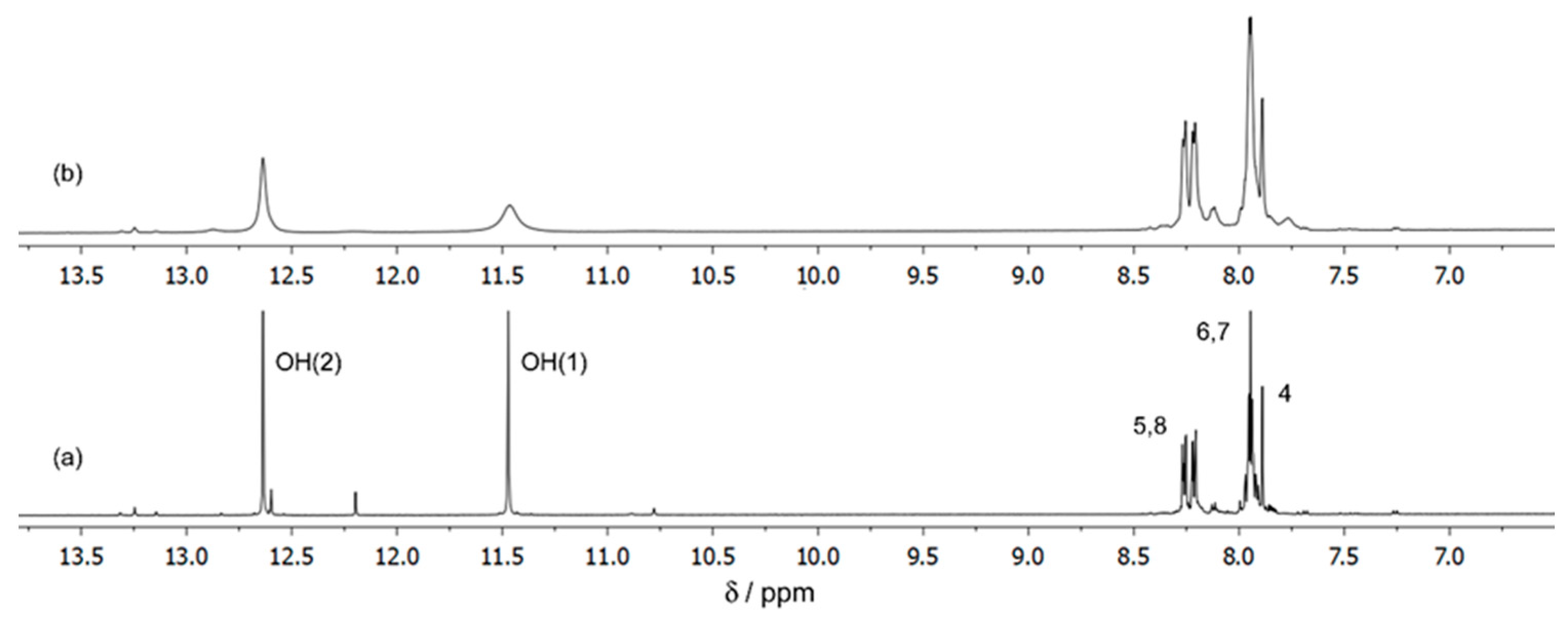
3.2.1. NMR Studies on the Ga(III)/ARS System
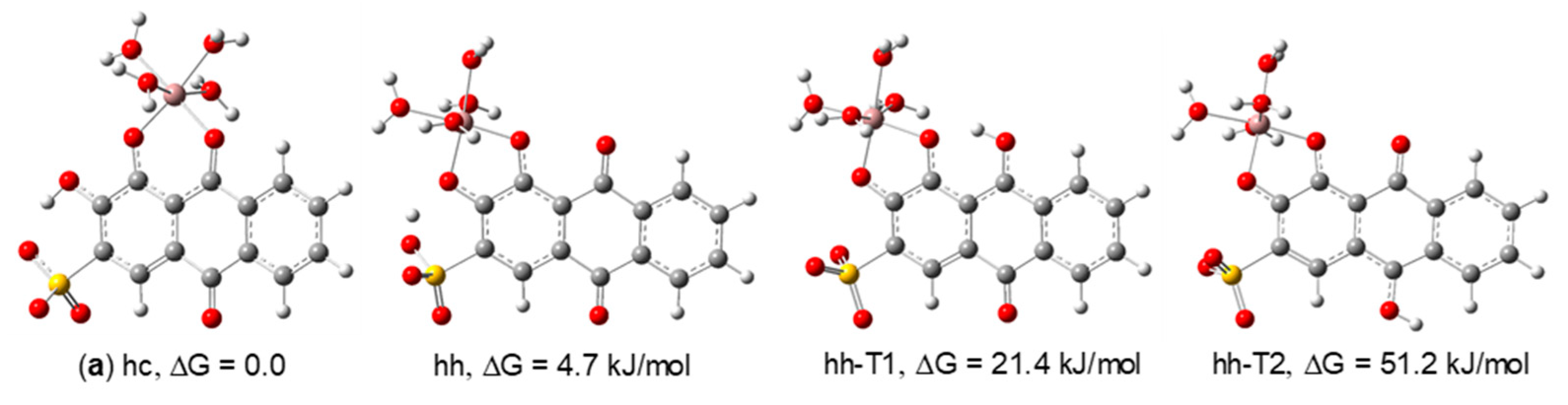
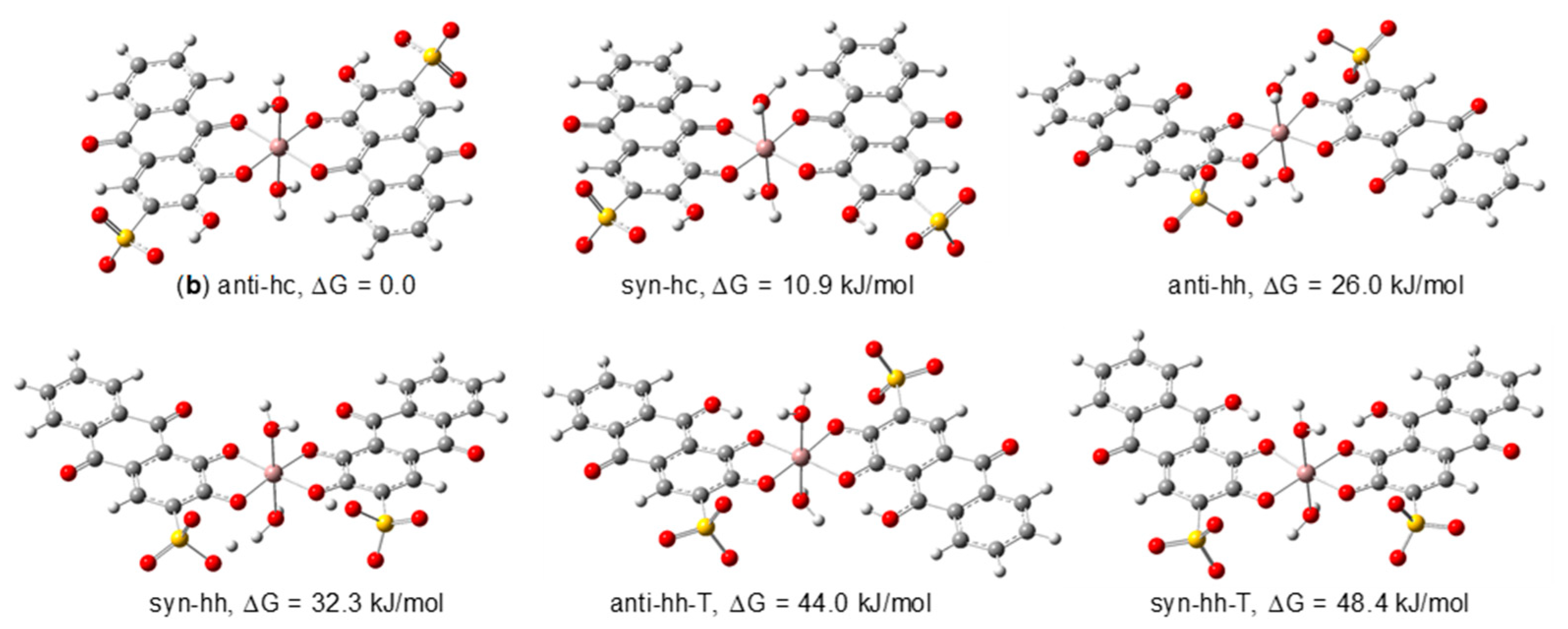
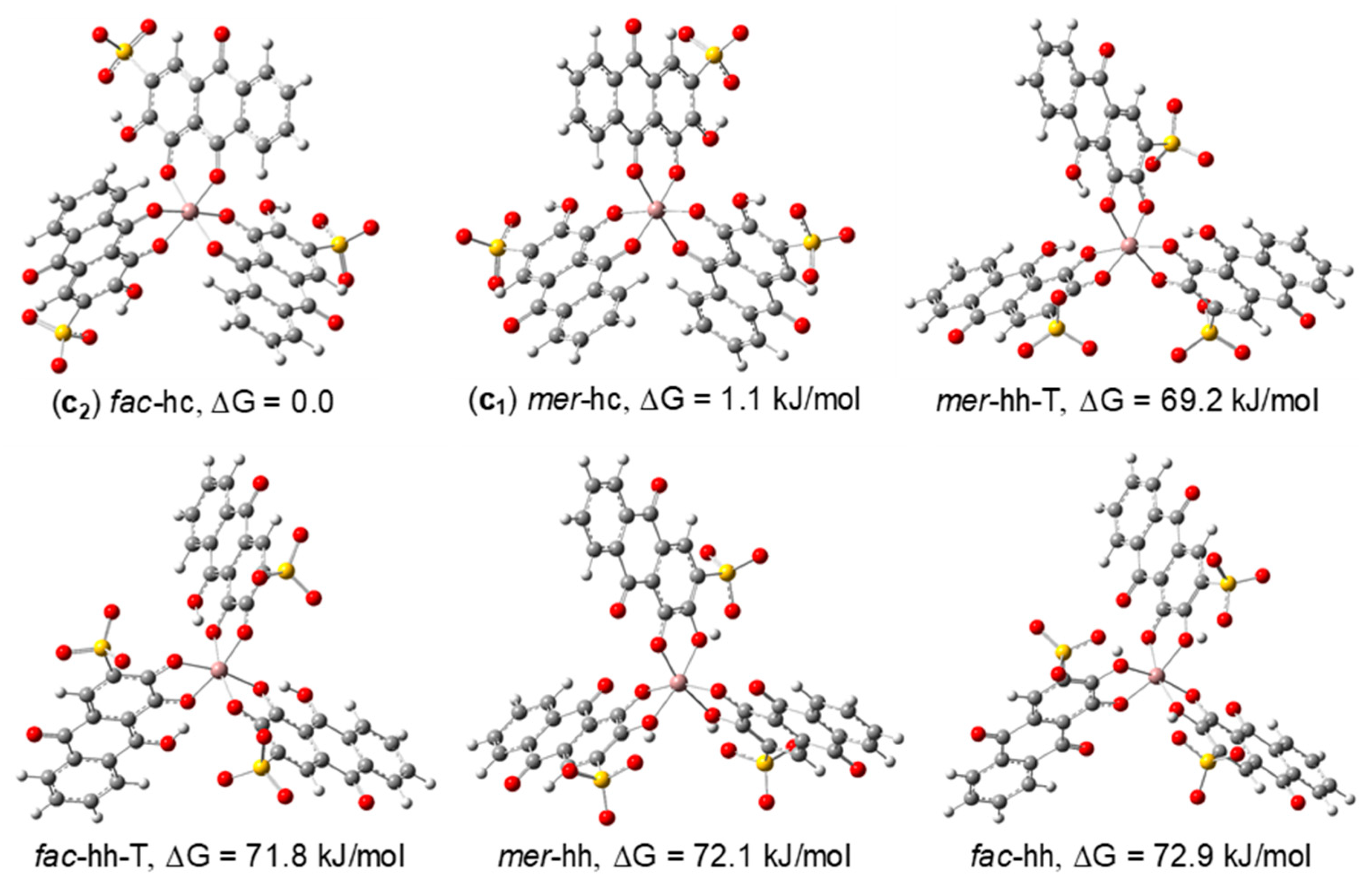
3.2.2. DFT Structural Characterization of the Ga(III)/ARS Complexes
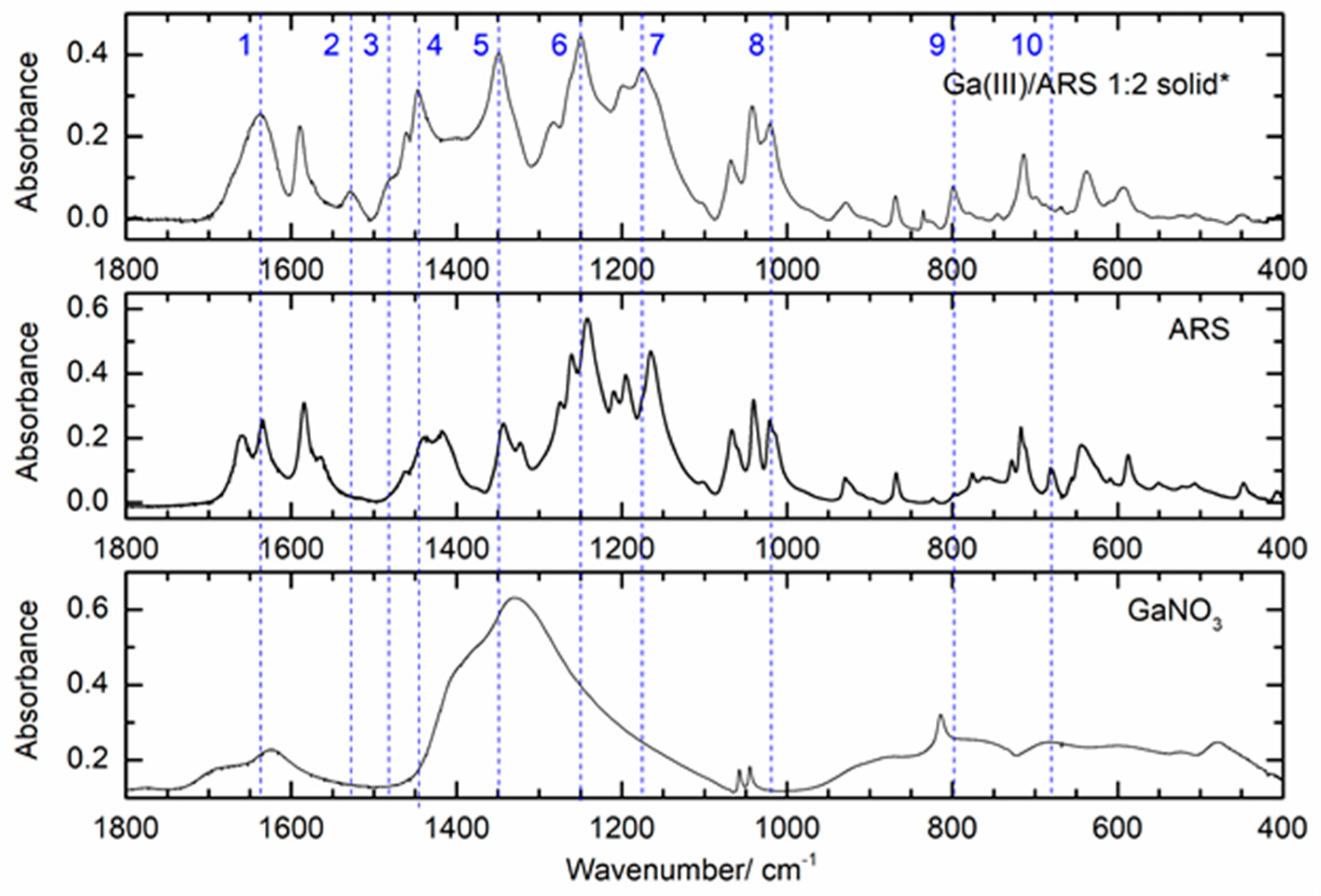
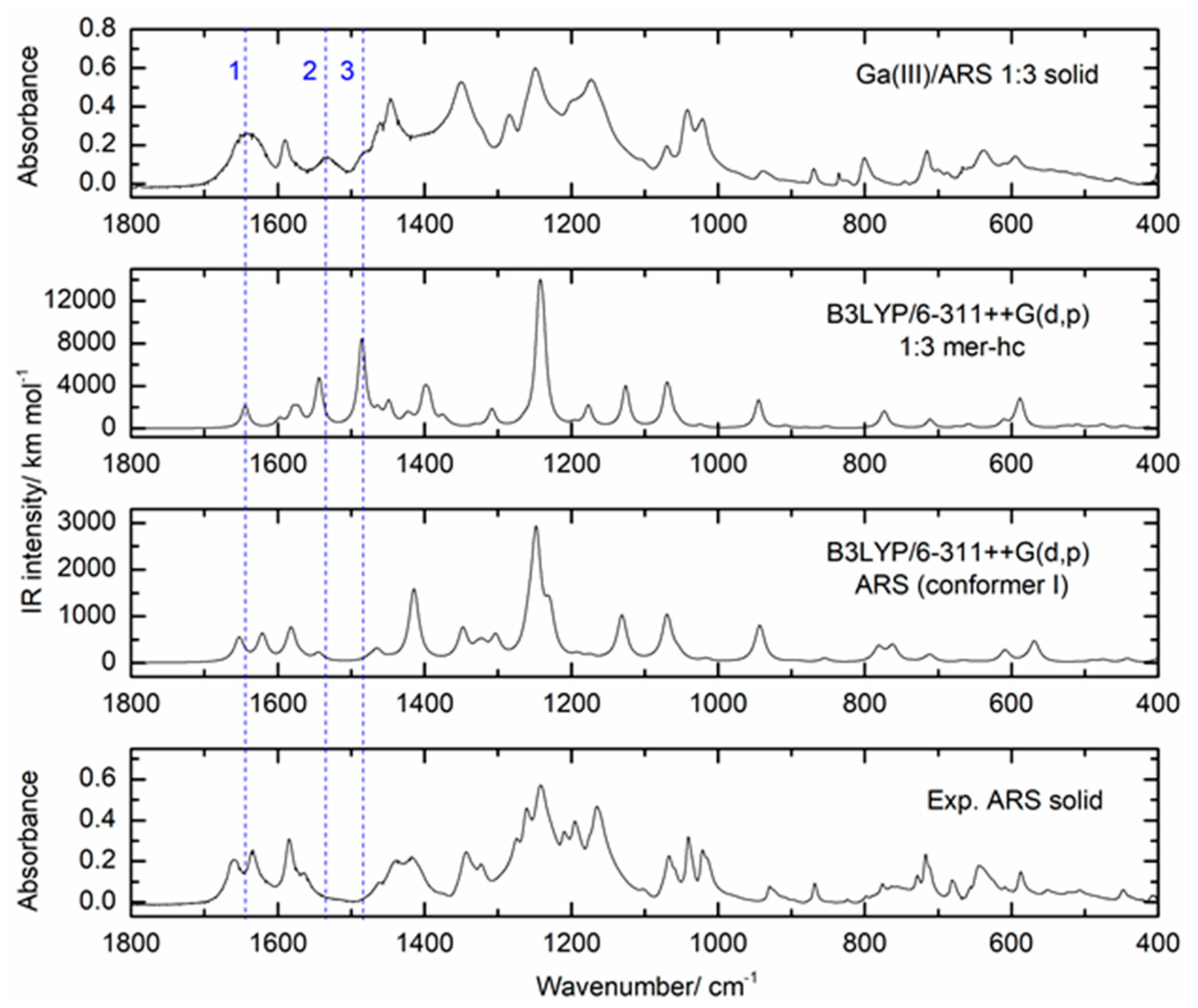
3.2.3. ATR-FTIR Studies on the Ga(III)/ARS System
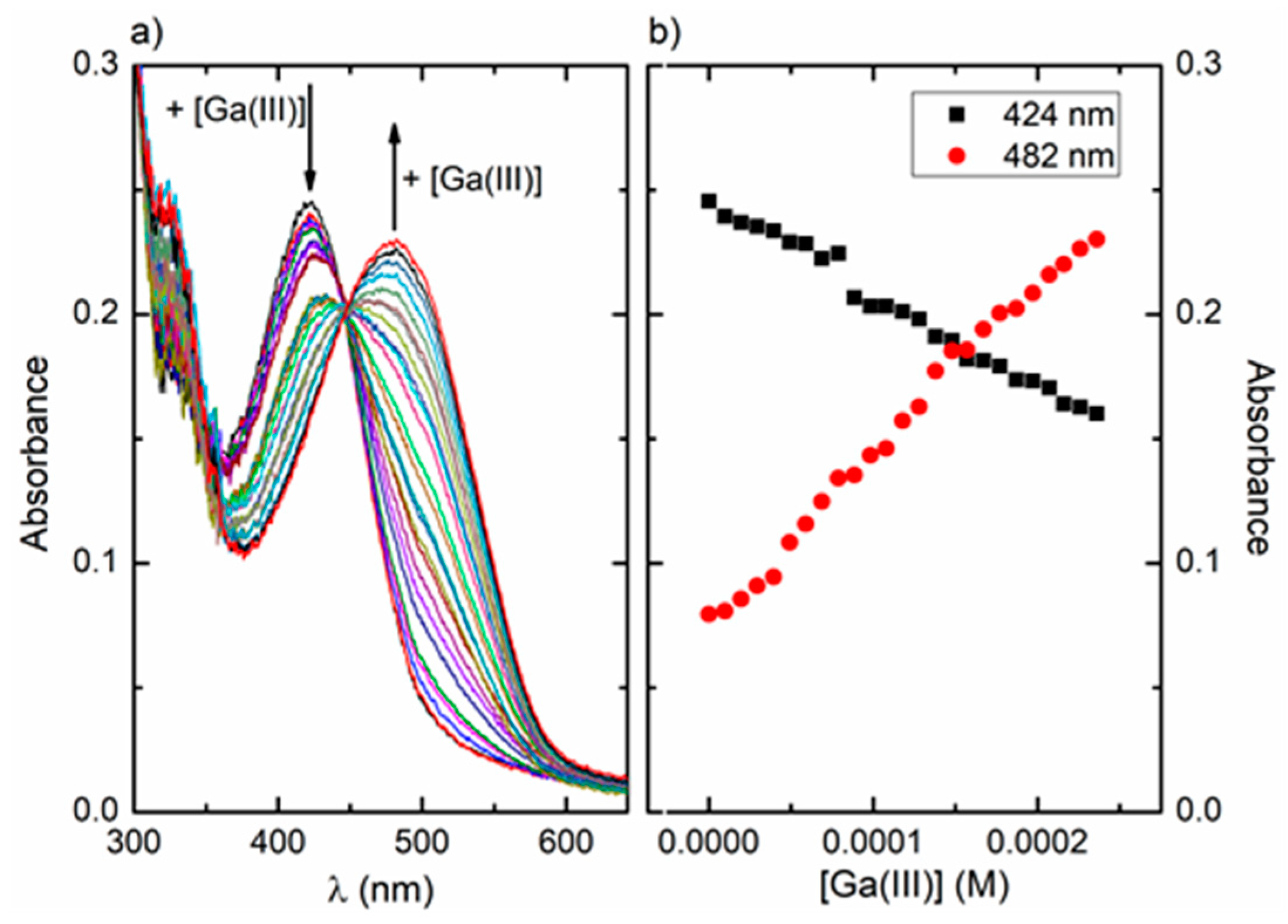
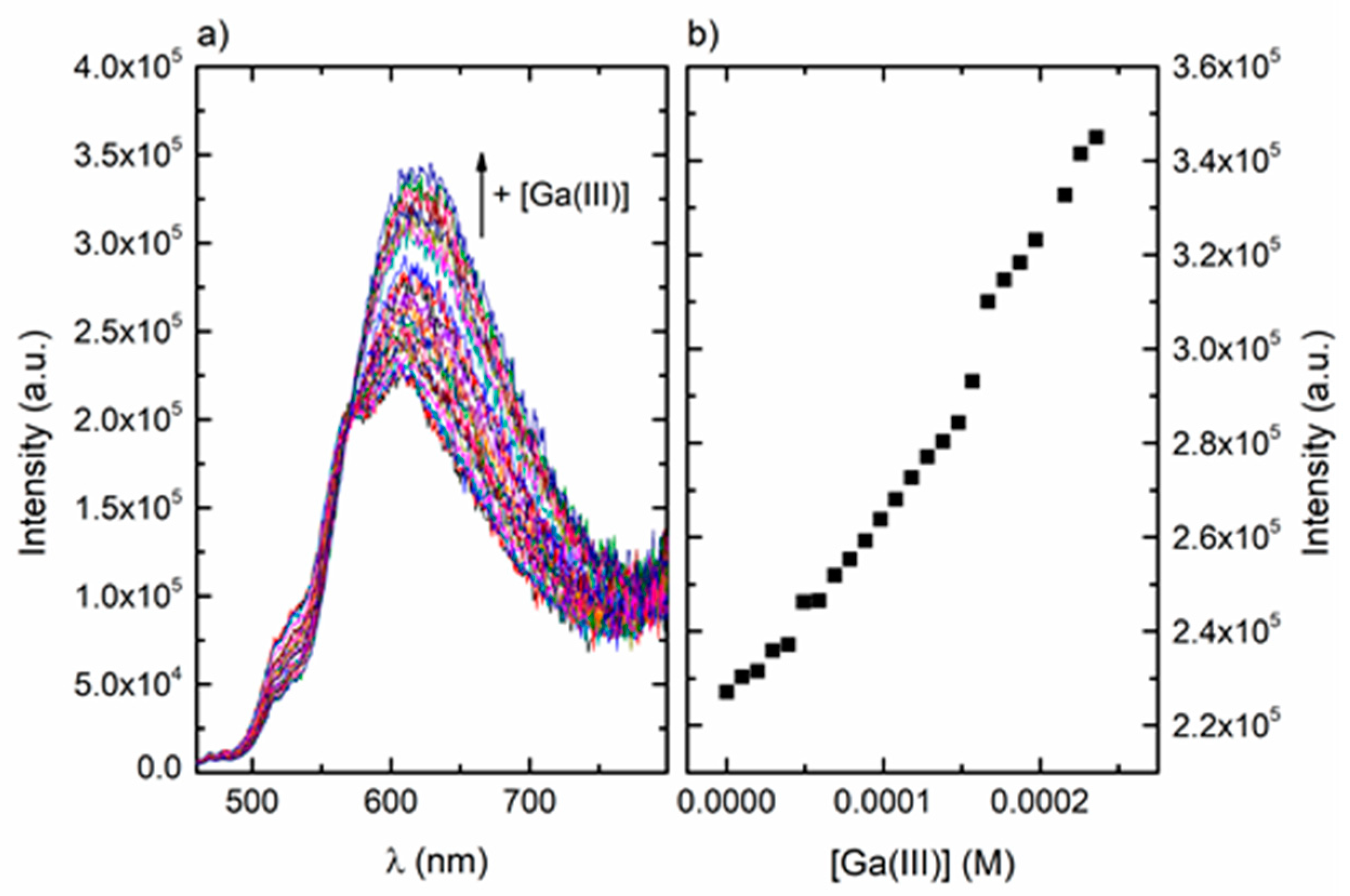
3.2.4. UV-Visible Absorption and Fluorescence Studies on the Ga(III)/ARS System
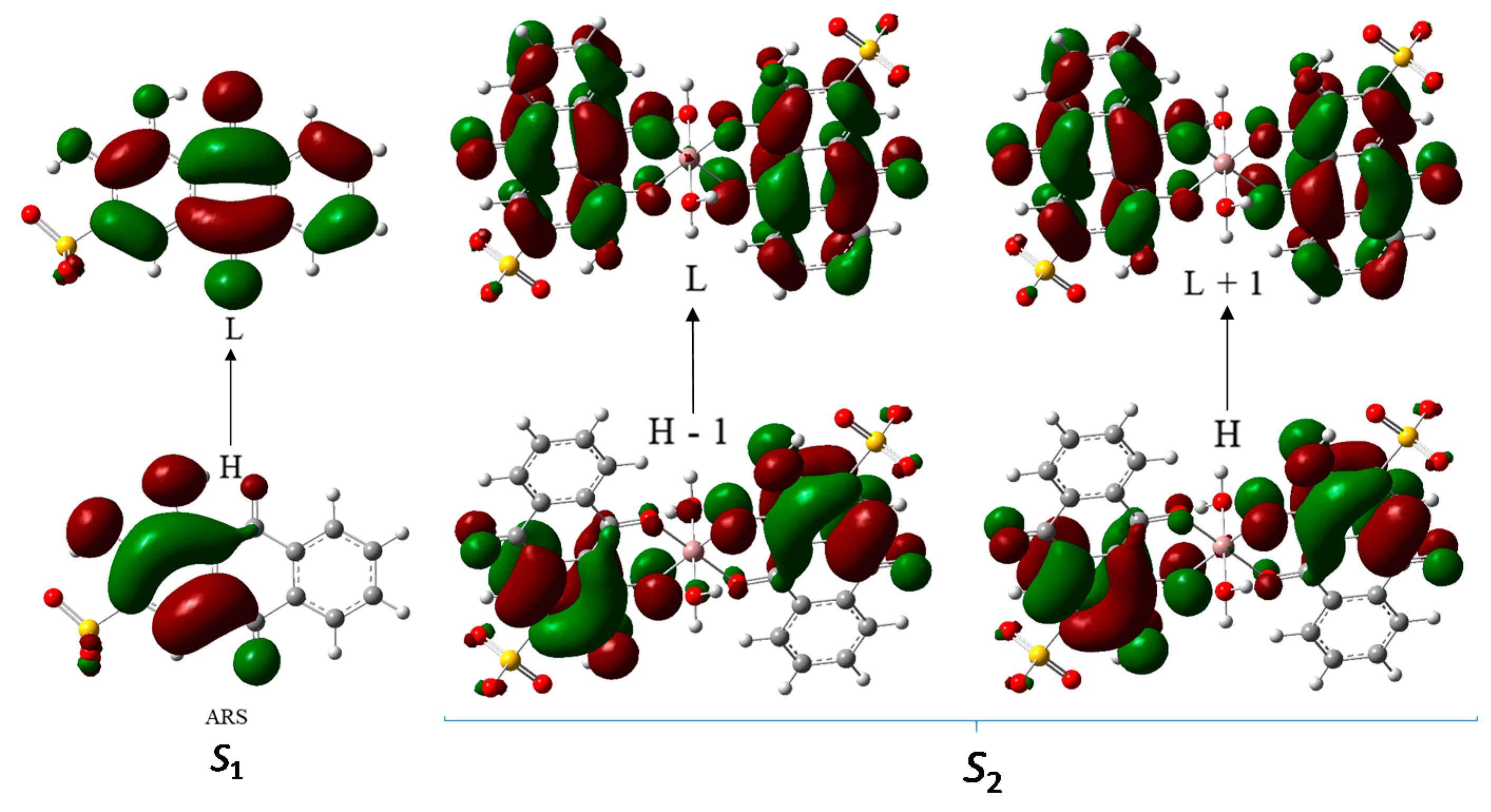
3.2.5. TD-DFT Studies on the Ga(III)/ARS System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sato, T.; Imai, M. Characteristics of nitrogen-doped GaAsP light-emitting diodes. Jpn. J. Appl. Phys. 2002, 41, 5995–5998. [Google Scholar] [CrossRef]
- Borra, E.F.; Tremblay, G.; Huot, Y.; Gauvin, J. Gallium liquid mirrors: Basic technology, optical-shop tests and observations. Astron. Soc. Pac. 1997, 109, 319–325. [Google Scholar] [CrossRef]
- Chua, M.-S.; Bernstein, L.R.; Li, R.; So, S.K.S. Gallium maltolate is a promising chemotherapeutic agent for the treatment of hepatocellular carcinoma. Anticancer Res. 2006, 26, 1739–1744. [Google Scholar] [PubMed]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartingerand, C.C.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalton Trans. 2008, 2, 183–194. [Google Scholar] [CrossRef]
- Baran, E.J. La nueva farmacoterapia inorgánica XIX. Compuestos de galio. Latin Am. J. Pharm. 2008, 27, 776–779. [Google Scholar]
- Chen, H.-W. Gallium, indium, and arsenic pollution of groundwater from a semiconductor manufacturing area of taiwan. Bull. Environ. Contam. Toxicol. 2006, 77, 289–296. [Google Scholar] [CrossRef]
- Powe, A.M.; Das, S.; Lowry, M.; El-Zahab, B.; Fakayode, S.O.; Geng, M.L.; Baker, G.A.; Wang, L.; McCarroll, M.E.; Patonay, G.; et al. Molecular fluorescence, phosphorescence, and chemiluminescence spectrometry. Anal. Chem. 2010, 82, 4865–4894. [Google Scholar] [CrossRef]
- Novotná, P.; Pacáková, V.; Bosáková, Z.; Štulík, K. High-performance liquid chromatographic determination of some anthraquinone and naphthoquinone dyes occurring in historical textiles. J. Chromatogr. A 1999, 863, 235–241. [Google Scholar] [CrossRef]
- Orska-Gawryś, J.; Surowiec, I.; Kehl, J.; Rejniak, H.; Urbaniak-Walczak, K.; Trojanowicz, M. Identification of natural dyes in archeological Coptic textiles by liquid chromatography with diode array detection. J. Chromatogr. A 2003, 989, 239–248. [Google Scholar] [CrossRef]
- Szostek, B.; Orska-Gawrys, J.; Surowiec, I.; Trojanowicz, M. Investigation of natural dyes occurring in historical Coptic textiles by high-performance liquid chromatography with UV–Vis and mass spectrometric detection. J. Chromatogr. A 2003, 1012, 179–192. [Google Scholar] [CrossRef]
- Alves, D.S.; Perez-Fons, L.; Estepa, A.; Micol, V. Membrane-related effects underlying the biological activity of the anthraquinones emodin and barbaloin. Biochem. Pharmacol. 2004, 68, 549–561. [Google Scholar] [CrossRef]
- Norton, S.A. Useful plants of dermatology. IV. Alizarin red and madder. J. Am. Acad. Dermatol. 1998, 39, 484–485. [Google Scholar] [CrossRef]
- Diaz, A.N. Analytical applications of 1,10-anthraquinones: A review. Talanta 1991, 38, 571–588. [Google Scholar] [CrossRef]
- Ghosh, A.; Jose, D.A.; Kaushik, R. Anthraquinones as versatile colorimetric reagent for anions. Sens. Actuators B Chem. 2016, 229, 545–560. [Google Scholar] [CrossRef]
- Diaz, A.N. Absorption and emission spectroscopy and photochemistry of 1,10-anthraquinone derivatives: A review. J. Photochem. Photobiol. A Chem. 1990, 53, 141–167. [Google Scholar] [CrossRef]
- Miliani, C.; Romani, A.; Favaro, G. Acidichromic effects in 1,2-di- and 1,2,4-trihydroxyanthraquinones. A spectrophotometric and Fuorimetric study. J. Phys. Org. Chem. 2000, 13, 141–150. [Google Scholar] [CrossRef]
- Duncan, W.R.; Prezhdo, O.V. Temperature independence of the photoinduced electron injection in dye-sensitized TiO2 rationalized by ab initio time-domain density functional theory. J. Am. Chem. Soc. 2008, 130, 9756–9762. [Google Scholar] [CrossRef]
- Kaniyankandy, S.; Verma, S.; Mondal, J.A.; Palit, D.K.; Ghosh, H.N. Evidence of multiple electron injection and slow back electron transfer in alizarin-sensitized ultrasmall TiO2 particles. J. Phys. Chem. C 2009, 113, 3593–3599. [Google Scholar] [CrossRef]
- Nawrocka, A.; Krawczyk, S. Electronic excited state of alizarin dye adsorbed on TiO2 nanoparticles: A study by electroabsorption (stark effect) spectroscopy. J. Phys. Chem. C 2008, 112, 10233–10241. [Google Scholar] [CrossRef]
- Lima, A.R.F.; Pereira, R.C.; Azevedo, J.; Mendes, A.; de Melo, J.S.S. On the path to aqueous organic redox flow batteries: Alizarin red S alkaline negolyte. Performance evaluation and photochemical studies. J. Mol. Liq. 2021, 336, 116364. [Google Scholar] [CrossRef]
- Safavi, A.; Abdollahi, H.; Mirzajani, R. Simultaneous spectrophotometric determination of Fe(III), Al(III) and Cu(II) by partial least-squares calibration method. Spectrochim. Acta Part A 2006, 63, 196–199. [Google Scholar] [CrossRef]
- Sathish, R.S.; Kumar, M.R.; Rao, G.N.; Kumar, K.A.; Janardhana, C. A water-soluble fluorescent fluoride ion probe based on Alizarin Red S–Al(III) complex. Spectrochim. Acta Part A 2007, 66, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, L.; Geng, F.; Zhang, F.; Xu, M. Design of a dual-signaling sensing system for fluorescent ratiometric detection of Al3+ ion based on the inner-filter effect. Analyst 2011, 136, 4809. [Google Scholar] [CrossRef] [PubMed]
- Supian, S.M.; Ling, T.L.; Heng, L.Y.; Chong, K.F. Quantitative determination of Al(III) ion by using alizarin red S including its microspheres optical sensing Material. Anal. Methods 2013, 5, 2602–2609. [Google Scholar] [CrossRef]
- Epstein, M.; Yariv, S. Visible-spectroscopy study of the adsorption of alizarinate by Al-montmorillonite in aqueous suspensions and in solid state. J. Colloid Interface Sci. 2003, 263, 377–385. [Google Scholar] [CrossRef]
- Biver, T.; Kraiem, M.; Secco, F.; Venturini, M. On the mechanism of indium(III) complex formation with metallochromic indicators. Polyhedron 2018, 156, 6–13. [Google Scholar] [CrossRef]
- Dwivedi, C.D.; Munshi, K.N.; Dey, A.K. Chelate formation of trivalent gallium with 1,2-dihydroxy-3-anthraquinone sulfonic acid. Microchem. J. 1965, 9, 218–226. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Perdew, J.P. Electronic Structure of Solids ’91; Ziesche, P., Eschrig, H., Eds.; Akademie Verlag: Berlin, Germany, 1991; Volume 11. [Google Scholar]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Erratum: Atoms, molecules, solids, and surfaces–Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Burke, K.; Perdew, J.P.; Wang, Y. Derivation of a Generalized Gradient Approximation: The PW91 Density Functional. In Electronic Density Functional Theory: Recent Progress and New Directions; Dobson, J.F., Vignale, G., Das, M.P., Eds.; Plenum Press: New York, NY, USA, 1998; pp. 81–111. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Justino, L.L.G.; Reva, I.; Fausto, R. Thermally and vibrationally induced conformational isomerizations, infrared spectra, and photochemistry of gallic acid in low-temperature matrices. J. Chem. Phys. 2016, 145, 014304. [Google Scholar] [CrossRef] [Green Version]
- Jaquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Shalaby, A.A.; Mohamed, A.A. Determination of acid dissociation constants of alizarin red S, methyl orange, bromothymol blue and bromophenol blue using a digital camera. RSC Adv. 2020, 10, 11311–11316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehér, P.P.; Purgel, M.; Joó, F. Performance of exchange–correlation functionals on describing ground state geometries and excitations of alizarin red S: Effect of complexation and degree of deprotonation. Comput. Theor. Chem. 2014, 1045, 113–122. [Google Scholar] [CrossRef]
- Ramos, M.L.; Justino, L.L.G.; Salvador, A.I.N.; de Sousa, A.R.E.; Abreu, P.E.; Fonseca, S.M.; Burrows, H.D. NMR, DFT and luminescence studies of the complexation of Al(III) with 8-hydroxyquinoline-5-sulfonate. Dalton Trans. 2012, 41, 12478–12489. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.L.; Justino, L.L.G.; de Sousa, A.R.E.; Fonseca, S.M.; Geraldes, C.F.G.C.; Burrows, H.D. Structural and photophysical studies on gallium(III) 8-hydroxyquinoline-5-sulfonates. Does excited state decay involve ligand photolabilisation? Dalton Trans. 2013, 42, 3682–3694. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.L.; Justino, L.L.G.; Fonseca, S.M.; Burrows, H.D. NMR, DFT and luminescence studies of the complexation of V(V) oxoions in solution with 8-hydroxyquinoline-5-sulfonate. New J. Chem. 2015, 39, 1488–1497. [Google Scholar] [CrossRef]
- Ramos, M.L.; Justino, L.L.G.; Abreu, P.E.; Fonseca, S.M.; Burrows, H.D. Oxocomplexes of Mo(VI) and W(VI) with 8-hydroxyquinoline-5-sulfonate in solution: Structural studies and the effect of the metal ion on the photophysical behavior. Dalton Trans. 2015, 44, 19076–19089. [Google Scholar] [CrossRef]
- Caldeira, M.M.; Ramos, M.L.; Cavaleiro, A.M.; Gil, V.M.S. Multinuclear NMR study of vanadium(V) complexation with tartaric and citric acids. J. Mol. Struct. 1988, 174, 461–466. [Google Scholar] [CrossRef]
- Ramos, M.L.; Caldeira, M.M.; Gil, V.M.S. NMR study of the complexation of D-galactonic acid with tungsten(VI) and molybdenum(VI). Carbohydr. Res. 1997, 297, 191–200. [Google Scholar] [CrossRef]
- Ramos, M.L.; Caldeira, M.M.; Gil, V.M.S. Multinuclear NMR study of complexation of D-galactaric and D-mannaric acids with tungsten(VI) oxoions. J. Coord. Chem. 1994, 33, 319–329. [Google Scholar] [CrossRef]
- Ramos, M.L.; Caldeira, M.M.; Gil, V.M.S. NMR spectroscopy study of the complexation of L-mannonic acid with tungsten(VI) and molybdenum(VI). Carbohydr. Res. 1997, 299, 209–220. [Google Scholar] [CrossRef]
- Ramos, M.L.; Caldeira, M.M.; Gil, V.M.S. NMR spectroscopy study of the complexation of D-gluconic acid with tungsten(VI) and molybdenum(VI). Carbohydr. Res. 1997, 304, 97–109. [Google Scholar] [CrossRef]
- Ramos, M.L.; Pereira, M.M.; Beja, A.M.; Silva, M.R.; Paixão, J.A.; Gil, V.M.S. NMR and X-ray diffraction studies of the complexation of D-(–)quinic acid with tungsten(VI) and molybdenum(VI). J. Chem. Soc. Dalton Trans. 2002, 10, 2126–2131. [Google Scholar] [CrossRef]
- Ramos, M.L.; Caldeira, M.M.; Gil, V.M.S. Multinuclear NMR study of the complexation of D-glucaric acid with molybdenum(VI) and tungsten(VI). Inorg. Chim. Acta 1991, 180, 219–224. [Google Scholar] [CrossRef]
- Justino, L.L.G.; Ramos, M.L.; Kaupp, M.; Burrows, H.D.; Fiolhais, C.; Gil, V.M.S. Density functional theory study of the oxoperoxo vanadium(V) complexes of glycolic acid. Structural correlations with NMR chemical shifts. Dalton Trans. 2009, 44, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Justino, L.L.G.; Ramos, M.L.; Nogueira, F.; Sobral, A.J.F.N.; Geraldes, C.F.G.C.; Kaupp, M.; Burrows, H.D.; Fiolhais, C.; Gil, V.M.S. Oxoperoxo vanadium(V) complexes of L-lactic acid: Density functional theory study of structure and NMR chemical shifts. Inorg. Chem. 2008, 47, 7317–7326. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, L.R.; Tanner, T.; Godfrey, C.; Noll, B. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Met.-Based Drugs 2000, 7, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Enyedy, É.A.; Dömötör, O.; Varga, E.; Kiss, T.; Trondl, R.; Hartinger, C.G.; Keppler, B.K. Comparative solution equilibrium studies of anticancer gallium(III) complexes of 8-hydroxyquinoline and hydroxy(thio)pyrone ligands. J. Inorg. Biochem. 2012, 117, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Lima, C.F.R.A.C.; Taveira, R.J.S.; Costa, J.C.S.; Fernandes, A.M.; Melo, A.; Silva, A.M.S.; Santos, L.M.N.B.F. Understanding M–ligand bonding and mer-/fac isomerism in tris(8-hydroxyquinolinate) metallic complexes. Phys. Chem. Chem. Phys. 2016, 18, 16555–16565. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.C.S.; Lima, C.F.R.A.C.; Santos, L.M.N.B.F. Electron transport materials for organic light-emitting diodes: Understanding the crystal and molecular stability of the tris(8-hydroxyquinolines) of Al, Ga, and In. J. Phys. Chem. C 2014, 118, 21762–21769. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | I | II | III | IV | V | VI |
|---|---|---|---|---|---|---|
| Symmetry | C1 | C1 | Cs | C1 | Cs | Cs |
| ΔEel | 0.0 | 18.8 | 26.0 | 40.7 | 71.8 | 73.6 |
| Δ(ETotal) | 0.0 | 18.1 | 24.8 | 38.1 | 70.4 | 71.0 |
| ΔG298K | 0.0 | 15.9 | 19.3 | 36.9 | 60.3 | 62.0 |
| P(%)298 | 99.8 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 |
| δ 1H RMN (exp.) a (H4) | Relative Concentration (%) | δ 1H RMN (exp.) a (H4) | Relative Concentration (%) | ||
|---|---|---|---|---|---|
| Temp. 280.15 K | Temp. 298.15 K | ||||
| c1 | 8.07 (I) | 57.5 | c1 | 8.04 (I) | 51.4 |
| 8.08 (II) | 8.05 (II) | ||||
| 8.11 (III) | 8.10 (III) | ||||
| c2 | 8.14 | 17.2 | c2 | 8.14 (broad) | 24.0 |
| Free lig. | 7.96 | 25.3 | Free lig. | 7.96 | 24.6 |
| M:L | Structure a | Sym | B3LYP | CAM-B3LYP | B3PW91 | w-B97X-D | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔG298 | P298 | ΔG298 | P298 | ΔG298 | P298 | ΔG298 | P298 | |||
| 1:1 | hc (a) | C1 | 0.0 | 86.8 | 0.0 | 69.8 | 0.0 | 77.6 | 0.0 | 52.2 |
| hh | C1 | 4.7 | 13.1 | 2.1 | 30.2 | 3.1 | 22.4 | 0.2 | 47.8 | |
| hh-T1 | C1 | 21.4 | 0.0 | 21.7 | 0.0 | 18.3 | 0.0 | 23.7 | 0.0 | |
| hh-T2 | C1 | 51.2 | 0.0 | 58.8 | 0.0 | 51.0 | 0.0 | 58.1 | 0.0 | |
| 1:2 | anti-hc (b) | Ci | 0.0 | 98.8 | 0.0 | 96.7 | 0.0 | 97.5 | 0.0 | 87.3 |
| syn-hc | Csb | 10.9 | 1.2 | 8.3 | 3.3 | 9.1 | 2.5 | 4.8 | 12.7 | |
| anti-hh | Ci | 26.0 | 0.0 | 27.1 | 0.0 | 24.3 | 0.0 | 23.0 | 0.0 | |
| syn-hh | Csb | 32.3 | 0.0 | 30.5 | 0.0 | 28.9 | 0.0 | 36.9 | 0.0 | |
| anti-hh-T | Ci | 44.0 | 0.0 | 53.5 | 0.0 | 40.2 | 0.0 | 51.8 | 0.0 | |
| syn-hh-T | Csb | 48.4 | 0.0 | 58.1 | 0.0 | 44.1 | 0.0 | 58.0 | 0.0 | |
| 1:3 | fac-hc (c2) | C3b | 0.0 | 61.4 d | 0.0 | 76.7 | 12.2 c | 0.7 | 29.8 c | 0.0 |
| mer-hc (c1) | C1 | 1.1 | 38.6 d | 3.0 | 23.3 | 0.0 c | 99.3 | 0.0 c | 100 | |
| mer-hh | C1 | 72.1 | 0.0 | 63.9 | 0.0 | 73.4 c | 0.0 | 139.4 c | 0.0 | |
| mer-hh-T | C1 | 69.1 | 0.0 | 92.0 | 0.0 | 256.3 c | 0.0 | 316.6 c | 0.0 | |
| fac-hh | C3b | 72.9 | 0.0 | 67.2 | 0.0 | 78.5 c | 0.0 | 144.5 c | 0.0 | |
| fac-hh-T | C3b | 71.8 | 0.0 | 92.2 | 0.0 | 285.9 c | 0.0 | 204.7 c | 0.0 | |
| Excited State | Energy (eV) | λcalc. (nm) | λexp. (nm) | f | Major Contributions (%) | Character |
|---|---|---|---|---|---|---|
| ARS | ||||||
| S1 | 3.29 | 377 | 424 | 0.2267 | H→L (100%) | π → π* |
| 1:1 hc complex (a) | ||||||
| S1 | 2.78 | 446 | 482 | 0.1901 | H→L (100%) | π → π* |
| 1:2 anti-hc complex (b) | ||||||
| S1 | 2.84 | 436 | 482 | 0.0000 | H-1→L+1 (41%) + H→L (59%) | π → π* |
| S2 | 2.91 | 426 | 0.4307 | H-1→L (52%) + H→L+1 (48%) | π → π* | |
| 1:3 mer (c1) | ||||||
| S1 | 2.85 | 436 | 482 | 0.2675 | H-1→L (26%) + H-1→L+2 (20%) | π → π* |
| S3 | 2.90 | 427 | 0.4348 | H→L (24%) + H-1→L (21%) | π → π* | |
| 1:3 fac (c2) | ||||||
| S1 | 2.85 | 435 | 482 | 0.1532 | H-2→L (34%) + H→L+1 (22%) | π → π* |
| S2 | 2.85 | 435 | 0.1548 | H-1→L (33%) + H→L+2 (23%) | π → π* | |
| S3 | 2.91 | 426 | 0.4244 | H→L (46%) + H-1→L+2 (28%) | π → π* |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Justino, L.L.G.; Braz, S.; Ramos, M.L. Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution. Photochem 2023, 3, 61-81. https://doi.org/10.3390/photochem3010005
Justino LLG, Braz S, Ramos ML. Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution. Photochem. 2023; 3(1):61-81. https://doi.org/10.3390/photochem3010005
Chicago/Turabian StyleJustino, Licínia L. G., Sofia Braz, and M. Luísa Ramos. 2023. "Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution" Photochem 3, no. 1: 61-81. https://doi.org/10.3390/photochem3010005
APA StyleJustino, L. L. G., Braz, S., & Ramos, M. L. (2023). Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution. Photochem, 3(1), 61-81. https://doi.org/10.3390/photochem3010005