1. Introduction
Glaucoma is a group of progressive eye disorders that is characterized by damage to the optic nerve head because of the breakdown of retinal ganglion cells and their axons which merge to form the optic nerve. Glaucoma is the leading cause of irreversible blindness, with more than 80 million people estimated to be affected by the disease worldwide. This number is projected to exceed 110 million by 2040 [
1].
Glaucoma is associated with numerous risk factors including, but not limited to, age, gender, smoking history, race, and elevated intraocular pressure. Reports show a 36% greater prevalence of glaucoma in males than in females [
2]. Risk of developing glaucoma has also been shown to increase with age [
3]. Of individuals aged 40 years and above, 5.7% of Blacks have glaucoma whereas only 2.2% of white people do [
4]. It is worth noting that, perhaps due to lack of access to glaucoma screening and to treatment, glaucoma is often undiagnosed. Previous reports have indicated that more than 50% of people with glaucoma are undiagnosed [
5,
6]. In an effort to address this public health crisis, it is important to establish methods for screening patients for risk factors of glaucoma worldwide.
Glaucoma is strongly associated with increased intraocular fluid pressure (IOP), as the aqueous humor exerts increased force per area than usual on the internal surface of the eye. This may occur in glaucoma because of blockages in the trabecular meshwork (TM) of the eye, which normally functions to drain aqueous humor from the anterior chamber. Poor drainage results in the accumulation of aqueous humor and causes IOP elevation. Glaucomatous eyes with elevated intraocular pressure have been shown to exhibit abnormalities in both the extracellular matrices of the retina and lamina cribrosa, which result in decreased function of the optic nerve and therefore lower visual acuity [
7].
2. Glaucoma Pathogenesis
It is uncertain whether apoptosis of retinal ganglion cells (RGC) occurs due to dysfunction of optic axons at the optic nerve head or if the optic nerve dysfunction is due to RGC dysfunction and/or death. Does elevated IOP cause damage first at the RGC axons in the ONH or does the increase in pressure cause changes first in RGC somas leading to apoptosis?
Elevated IOP causes changes locally in the eye, so it would logically follow that retrobulbar areas would be less affected and would therefore show fewer changes. Examinations of retrobulbar areas in elevated IOP models support this assumption and have not shown similar extracellular accumulations that are seen in the eye [
8].
Elevated IOP has been suspected to induce negative effects by the formation of a hypoxic environment in the eye following a reduction in ocular blood flow (26). Glaucomatous neurodegeneration is often preceded by a reduction of ocular perfusion due to decreased blood flow. Additionally, not all patients with glaucoma have elevated intraocular pressure, exemplified by normal tension glaucoma. Ischemia and histopathological glaucomatous abnormalities may occur in conditions of elevated IOP as well as in normal IOP conditions [
9]. The commonality in these conditions is the degeneration of retinal ganglion cells.
Retinal ganglion cell somas are located in the inner surface of the retina. The axons of RGCs then course through the retinal nerve fiber layer and converge at the optic disc, where they bundle together to form the optic nerve. Studies on human patients have indicated that RGC somas and their axons in the retinal nerve fiber layer face damage simultaneously [
10,
11]. These findings postulate that the retina is the primary site of injury in glaucoma.
Conversely, early changes are often seen in the lamina cribrosa, a structure in the optic nerve head that is responsible for supporting the axons exiting the eye. The lamina cribrosa (LC), the mesh-like connective tissue in the optic nerve head, contains a meshwork of pores through which RGC nerve fibers pass through before converging to form the optic nerve. Along the optic nerve, supportive glial cells are present to provide cellular support for RGC axons and maintain communication between these cells. Astrocytes are commonly found in these locations and maintain RGC axon health by functioning in the extracellular space to maintain ion balances and, along with other maintenance functions, remodel the extracellular environment. In glaucomatous eyes, astrocyte function around the lamina cribrosa decreases which results in pathogenic remodeling of the extracellular matrix. These abnormalities, including extracellular depositions of collagens and basement membrane components, have been noted in pathologic studies of glaucomatous optic nerve heads in humans and monkeys [
12]. These abnormal depositions have been found in laminar pores that were previously occupied by clusters of RGC axons [
8]. The accumulation of these depositions can be observed clinically as optic cupping when evaluating cup-to-disc ratio (CDR). CDR is often used as a measure of glaucoma progression. A higher CDR occurs when the space taken up by the optic cup is a larger than normal percentage of the optic disc area. Elevated CDR indicates that there is increased empty space in the optic nerve head. While an elevated cup-to-disc ratio is often clinically observed in the early stages of glaucoma, it should not be assumed that glaucoma pathogenesis occurs in the optic nerve head. Pathological changes in retinal layers may precede ONH changes, but clinicians are currently limited in their ability to view retinal cell layers at the level at which changes occur. The future development and implementation of clinical tools to evaluate retinal cellular changes may provide further insight into glaucoma disease onset.
The portion of the optic nerve head that is supported by the lamina cribrosa appears to be particularly susceptible to changes in IOP. This area is unique as it is the only location in the central nervous system where RGC axons do not have direct contact with local capillaries and therefore are heavily reliant on surrounding astrocytes for maintaining cellular health [
13]. Isolated samples of human ONH cells from the lamina cribrosa have been demonstrated to express and secrete neurotrophic receptors, indicating one of their roles in retinal ganglion cell support [
14].
Mechanical disruption of the extracellular environment by increased IOP results in astrocyte hypertrophy, thereby impeding the maintenance of RGC axon health and leaving these axons vulnerable. Quigley et al. evaluated the eyes of glaucoma patients and found that axons were damaged in the lamina cribrosa. They concluded that the structural integrity of the lamina cribrosa is a factor in predicting susceptibility to elevated IOP [
15]. While lamina cribrosa abnormalities are certainly involved in the progression of glaucoma, definitive evidence suggesting that the disease originates in the ONH is lacking.
Retinal ganglion cell axons in the glaucomatous ONH exhibit several abnormalities which result in the degeneration of these axons. Studies have shown that in both animal models and humans, RGC axons in the ONH exhibit swelling. This axonal swelling has been proposed to represent the accumulation of transport vesicles, thereby demonstrating the dysfunctional retrograde and anterograde transport occurring in the ONH axons [
15,
16]. Inhibited transport may have many complications, including the prevention of neurotrophic factors from reaching the RGCs. Retinal ganglion cell axons eventually synapse on other neurons which release neurotrophic factor, indicating that the connection between these neurons is stable and therefore maintains the health of the neuronal connection. When such connections are disrupted, retinal ganglion cell axons and somas begin to degenerate. Morgan et al. showed that of all RGC axons passing through the lamina cribrosa, approximately 8–12% deviated to travel through a tight space between the collagenous plates [
17]. Therefore, these nerve fibers are more likely to be affected by slight increases in pressure as a result of elevated IOP. Increased pressure could potentially cause impaired neurotrophic transport to RGCs. However, there is conflicting evidence regarding the effects of elevated IOP on axonal transport deficits in the ONH. Abbot et al. evaluated axonal transport activity in rat glaucoma models after acute IOP exposure and found that IOP elevation did not result in a significant axonal transport deficit or loss of retinal ganglion cells [
18]. Furthermore, normal function of signal transduction of RGCs is not affected, as retinal ganglion cell soma produces both brain-derived neurotrophic factor and TrkB receptors themselves [
19].
3. Local Retinal Changes in Early Stages of Glaucoma
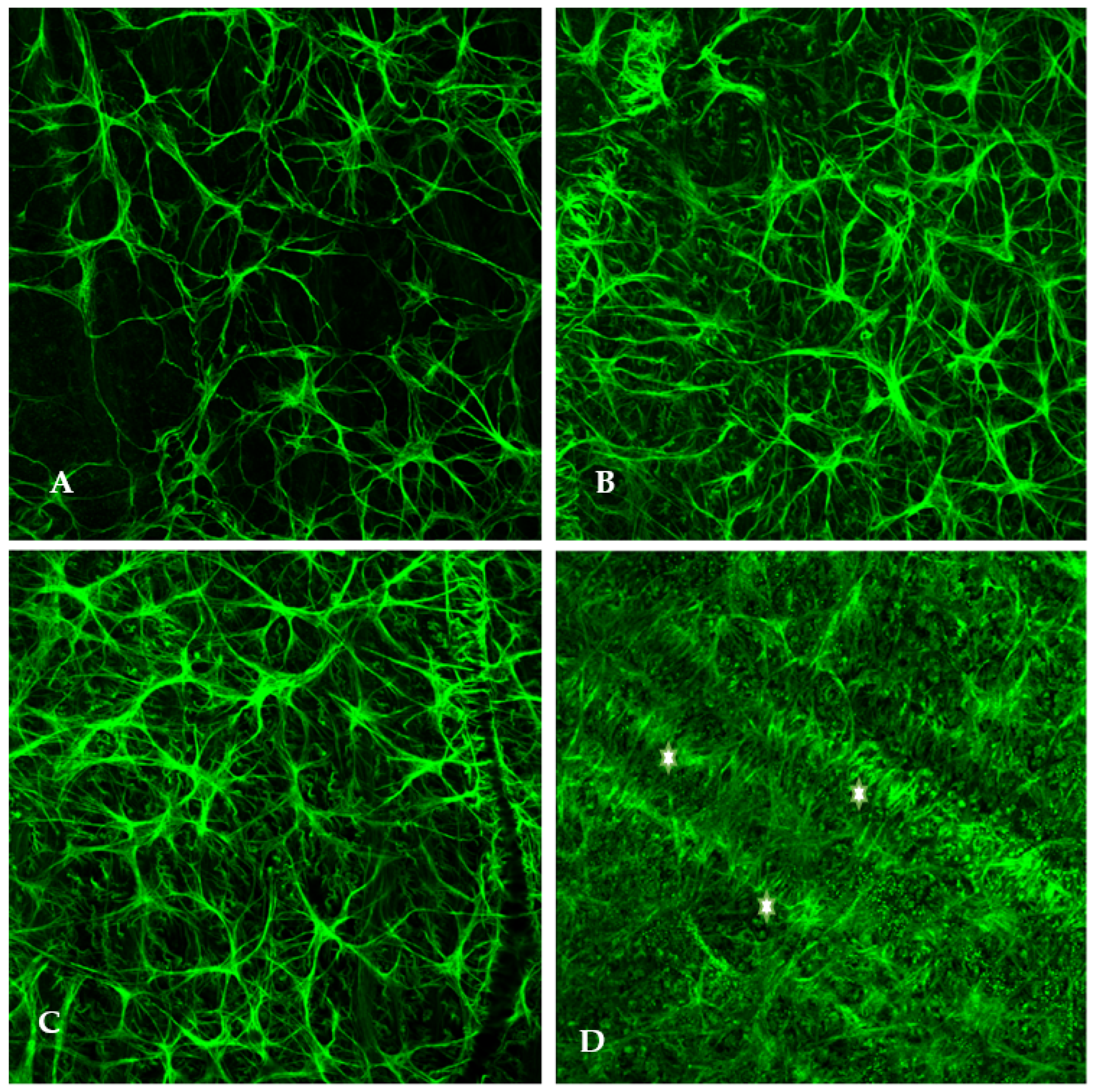
Elevated-IOP-induced damage is not mutually exclusive to the optic nerve head. As previously discussed, intraocular pressure elevation induces cellular stress which results in retinal astrocyte activation and hypertrophy. Using rat glaucoma models, we examined the retinal localization of astrocytic hypertrophy following the experimental elevation of IOP. Immunostaining of flat-mounted retinas was conducted using antibodies against glial fibrillary acidic protein (anti-GFAP), an intermediate filament protein expressed in astrocytes, to indicate the presence of normal and hypertrophied astrocytes (
Figure 1).
Within 48 h of elevated IOP (25 mm of Hg: normal IOP being 15–16 mmHg), retinal astrocytes begin to hypertrophy and their end feet on the blood vessels become increased in number and size (
Figure 1). This is a sign of hypoxia. Increased hypertrophy of astrocytes was observed parallel to the direction of optic axons. It is assumed that optic axons are being choked by the astrocytic processes.
Significantly, it is worth noting that optic disc cupping occurs several weeks after initial exposure to elevated IOP, but in the experimental animal model, we found that astrocyte hypertrophy was observed within the first 24–48 h following IOP elevation. Following the elevation of IOP, several changes were observed within one week including the hypertrophy of retinal astrocytes, overexpression of mutant superoxide dismutase, and osmotic imbalance via aquaporin 4 overexpression. Additionally, one can observe the increased presence of astrocytic end feet on the retinal vessels, indicating cellular stress. Astrocytes begin releasing ATP in the presence of retinal metabolic stress thereby leading to propagation of Ca++ waves and subsequent apoptosis.
As retinal ganglion cell somas face chronically elevated IOP and undergo stress-related changes, the health of their axons in the ONH will not be pertinent as the axons will undoubtedly degenerate. This further supports the notion that glaucomatous neurodegeneration begins in the retina and spreads to the optic nerve head. It is not necessarily astrocyte hypertrophy that is causing RGC axon death, but as shown, areas of high astrocyte density also contain large bundles of axons and strangulation remains a possibility. These results are supported by a study of astrocyte morphology in the early stages of glaucoma. Astrocytes begin sprouting smaller processes that invade areas near retinal ganglion cell axons [
20].
How does increased intraocular pressure cause damage to retinal ganglion cells? Intraocular pressure is maintained at about 10–20 mmHg above atmospheric pressure. Vision decline occurs when environmental exterior pressure changes during activities such as air traveling and diving [
21]. Transient receptor potential channel vanilloid 4 (TRPV4) has been implicated in a glutamate excitotoxicity mechanism of glaucomatous neurodegeneration. TRPV4, a receptor that is expressed in both mammalian bipolar cells and retinal ganglion cells, is sensitive to pressure changes. Pressure changes cause mechanical membrane movement which induces the opening of the TRPV4-linked cation channel [
22]. On retinal bipolar cells, TRPV4-induced cation influx likely increases glutamate release onto retinal ganglion cell synapses. This is supported by a study of Ryskamp et al., which demonstrated that with sustained exposure of mouse retinas to TRPV4 agonists, retinal ganglion cells exhibited dose-dependent apoptosis. Furthermore, studies have shown that TRPV4 increases both the rate at which retinal ganglion cells fire and the minimum stimulation the RGC must receive in order to fire [
22,
23]. TRPV4-mediated glutamate excitotoxicity of RGCs in response to elevated IOP provides another possible mechanism for glaucoma pathogenesis. Additionally, the pattern of vision loss in glaucoma aligns well with this mechanism. In the early stages of glaucoma vision loss, peripheral vision begins to worsen as a result of RGC death in the retinal periphery. Bipolar cells releasing glutamate may induce excitotoxicity in RGCs. In the peripheral areas of the retina that correspond to peripheral vision, retinal ganglion cells receive convergent inputs from several bipolar cells whereas there is less convergence of signals in the central retina [
24].
Glaucomatous degeneration can also be observed as the retinal nerve fiber layer decreases in thickness. RGC dysfunction and retinal nerve fiber layer thinning are directly correlated with glaucomatous visual field defects [
25]. Additionally, there is strong evidence suggesting that retinal tissue is one of the first tissues to be affected by intraocular pressure elevation. High-resolution angiograms have demonstrated that perfusion first becomes impaired in the retina before occurring in surrounding areas such as the choroid. Local perfusion, quantified as vascular area density, decreases first in the retina and reaches a level 50% of baseline at only 60 mmHg, whereas choroid vascular area density reaches 50% of baseline at 70 mmHg [
26].
4. The Role of Dysfunctional Retinal Mitochondria in Glaucoma
The retina is one of the most metabolically active tissues in the human body, making it particularly vulnerable when metabolic capacity and energy production are not ideal. Increased intraocular pressure results in decreased retinal metabolic function, which leaves RGC axons vulnerable and results in eventual apoptosis [
27,
28]. Retinal mitochondria, key cellular organelles involved in the axonal energetic support, become defective in glaucomatous models. Mitochondrial dysfunction is often attributed to the increased production of reactive oxygen species which interact with local molecules, such as mitochondrial DNA and cellular proteins, to inflict damage. As reactive oxygen species (ROS) accumulate and overwhelm the antioxidant capacity of the cell, a state of oxidative stress begins. Eventually, long-term exposure to oxidative stress induces apoptosis. Studies have indicated a strong correlation between the elevation of intraocular pressure and oxidative stress in retinal ganglion cell mitochondria [
28]. As intraocular pressure increases, a correlative increase in the quantity of oxidatively damaged DNA and RNA in retinal ganglion cells occurs [
28]. This data implicates reactive oxygen species generation in the pathogenesis of glaucomatous degeneration.
Munemasa et al. recently evaluated the role of thioredoxin, an antioxidant protein, in glaucoma models. Thioredoxin proteins function as neuroprotective free radical scavengers which neutralize ROS to prevent harmful oxidation of cellular components. The function of antioxidant proteins is inhibited by a protein called thioredoxin-interacting protein (TXNIP), thereby preventing ROS neutralization [
29]. Munemasa et al. noted increased expression of TXNIP in glaucomatous retinas, indicating that glaucomatous retinal cells are more vulnerable to ROS accumulation, and therefore more likely to be exposed to oxidative stress [
29]. With poor antioxidant defenses, the retinal ganglion cells’ redox state becomes irregular, and dysfunction ensues. Significantly, the upregulation of mitochondrial and cytosolic thioredoxin proteins resulted in increased survival of retinal ganglion cells affected by elevated intraocular pressure [
29].
What is the role of oxidative stress in the onset of glaucoma? It is important to consider possibilities such as the likelihood that disease origination occurs simultaneously in the retina and ONH. Increased age, genetic background of glaucoma, and elevated IOP are considered to be the leading risk factors for glaucoma [
11]. Therefore, among individuals of older age and individuals with familial history of glaucoma, it is expected that oxidative stress levels are elevated in the retina [
30]. In the aging retina, oxidative stress is one of the main causes of local tissue stress and subsequent inflammation. Acute exposure to oxidative stress results in the activation of normal inflammatory signaling but chronic exposure results in a detrimental inflammatory response [
31]. Local para-inflammation, initially existing in a homeostatic role in the retina, becomes dysregulated in glaucoma and has been linked to worsening trabecular meshwork dysfunction [
32]. Abnormal inflammatory signaling may also result in astrocyte activation and extracellular matrix remodeling. Evidence suggests that abnormal depositions of proteins and remodeling of the ECM result in increased stress in the endoplasmic reticulum of cells in the trabecular meshwork (TM) [
33]. These irregularities continue to worsen trabecular meshwork function and further contribute to TM blockages that cause elevated intraocular pressure.
It is critical to consider the significant role of astrocytes in glaucomatous neurodegeneration. As astrocytes continue to be activated as a result of dysregulated inflammatory signaling, they are no longer able to perform vital functions on a regular basis. Astrocytic activation may even cause direct harm to retinal ganglion cells through a glutamate excitotoxic mechanism. Studies have indicated that following astrocytic activation in glaucomatous eyes, glutamate transporters and receptors that are responsible for clearing the synaptic space become downregulated [
34]. Subsequent poor glutamate removal from the synapse may cause accumulation of the neurotransmitter on retinal ganglion cell dendrites which normally receive glutamate from bipolar cells. Long-term exposure to glutamate eventually causes apoptosis of these cells. Normal tension glaucoma, a variety of glaucoma in which intraocular pressures are not elevated yet glaucomatous optic neuropathy occurs, may offer further insight into the role of astrocytes in disease progression. Shinozaki et al. examined experimental knockout mice with deficient astrocyte transporters and found that these mice had normal IOP levels, but demonstrated age-associated retinal ganglion cell degeneration, thinning of retinal layers, axonal swelling, and impaired visual function [
35]. This study indicates that glaucomatous neurodegeneration and astrocyte defectivity may have a significant link. It is possible that astrocytic dysfunction is not involved in the pathogenesis of glaucoma but may heavily influence the rapid neurodegenerative aspect of the disease. Astrocytic activation and abnormal activity, as well as glial scarring in glaucoma, have been well documented. The similar activity of astrocytes is also observed in many neurodegenerative diseases such as Alzheimer’s Disease and Multiple Sclerosis, hence further research is needed to explore the exact nature of the role of astrocytes in glaucoma [
35].



{kind=link}
