3.1. Acid-Catalyzed Lignin Depolymerization
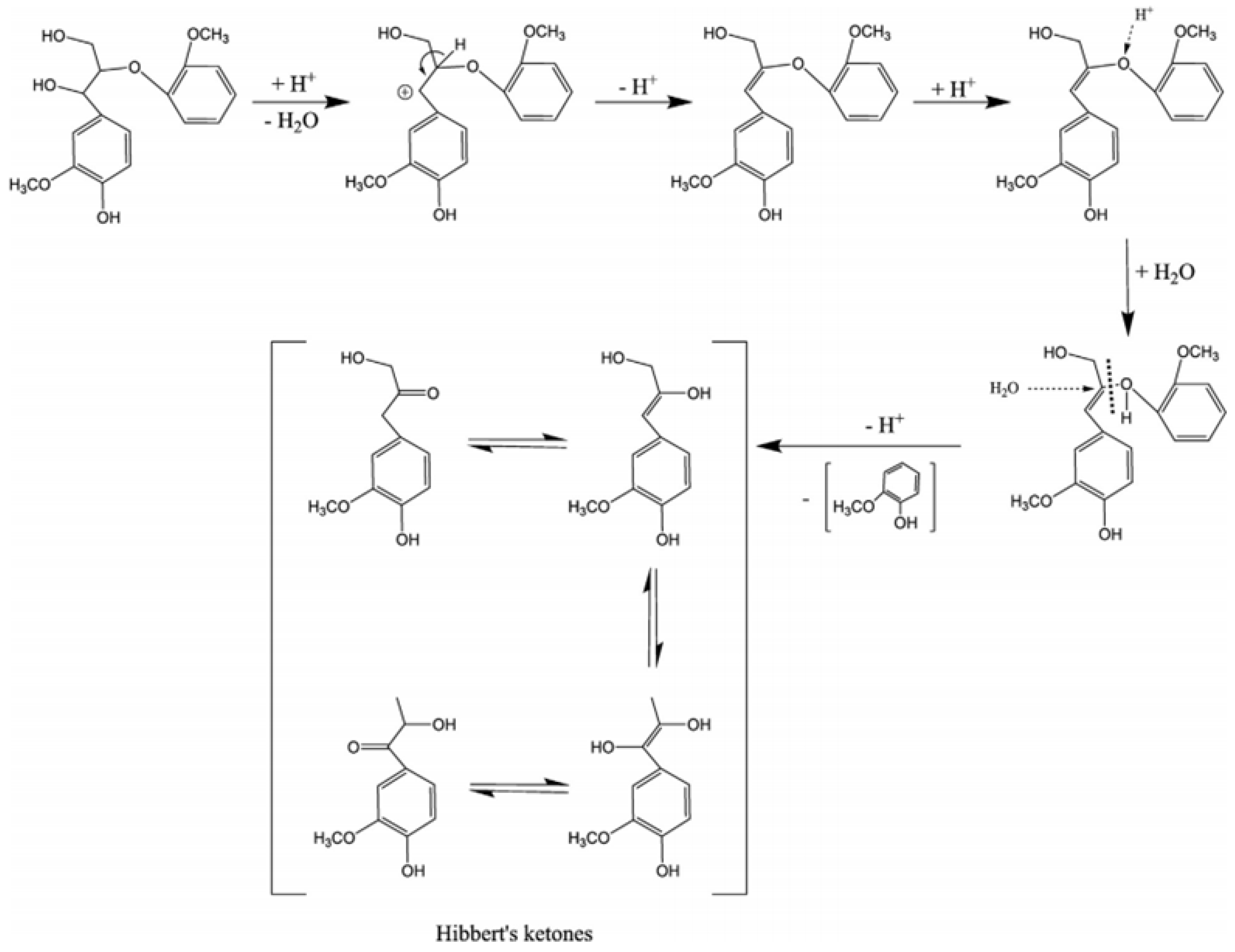
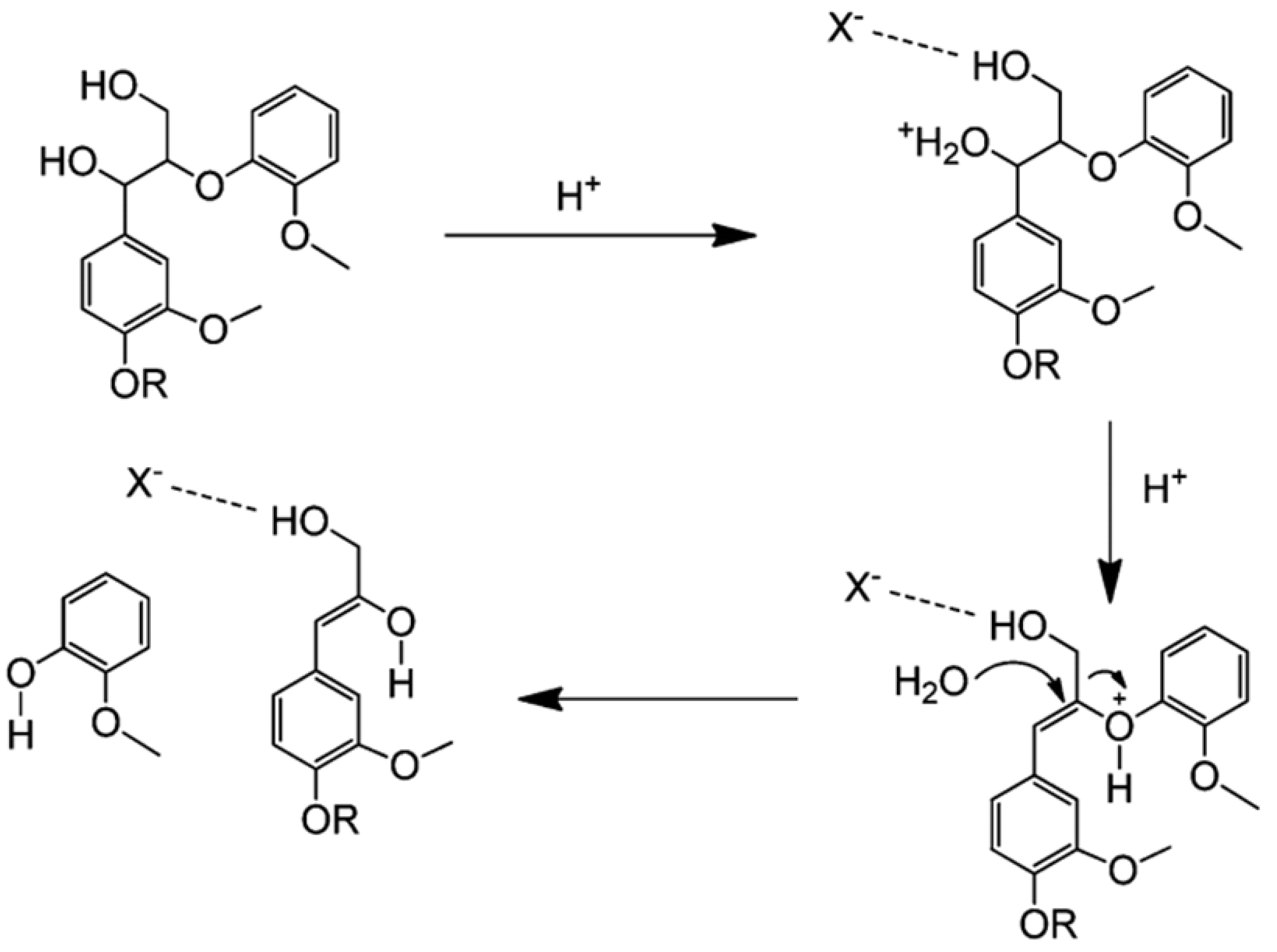
Depolymerization of lignin based on acid catalysis is a well-known method that has been used since the mid-twentieth century. This method decomposes the β-O-4 linkages of lignin using acid [
15]. Different acids, such as hydrochloric, sulfuric, formic, and peracetic, have been used as catalysts to break down the ether linkages [
51]. During hydrolysis, the acids provide H
3O
+ to break the ether bonds, producing phenolic compounds. An acid catalyst mechanism for the cleavage of β-O-4 is shown in
Figure 3.
These kinds of depolymerizations are usually conducted at high temperatures and pressures. A depolymerization study is typically performed using ethanol and acid by varying the solvent-acid ratio (e.g., hydrochloric acid/ethanol and formic acid/ethylene glycol) to separate the products into water-soluble and water-insoluble lignin. Mahmood et al. showed that a temperature range of 78–200 °C is insufficient to break the lignin structure into monomeric units [
52].
Figure 3.
Cleavage of β-O-4 linkages by acid catalysts, adapted from [
53].
Figure 3.
Cleavage of β-O-4 linkages by acid catalysts, adapted from [
53].
Recently, some research groups have investigated acid-catalyzed depolymerization at high temperatures and pressure using different proportions of acids [
54,
55]. One study suggested that a 10:77 (wt%) formic acid/ethanol mixture effectively depolymerized wheat straw lignin [
54]. Another group reported that 10:81 (wt%) of formic acid/ethanol is the optimal ratio to depolymerize lignin from wheat straw [
55]. Dilute sulfuric acid mixed with an ethanol/water solution was used for the depolymerization reaction, where the reaction achieved a high yield under 2 MPa and 250 °C for 1 h. Approximately 70 wt% of depolymerization was achieved when sulfuric acid was used as the catalyst in a 1:1 water/ethanol solvent system [
51]. Conditions for the acid-catalyzed depolymerization of lignin and the major outcomes are listed in
Table 2.
Although acid-catalyzed depolymerization is one of the most widely used methods, it has some disadvantages. Extreme reaction conditions, repolymerization, and toxic chemical use are unavoidable aspects of this method. Repolymerization mainly occurs between the reactive sites of the phenols and the α-carbon of phenol propanol [
66,
67]. This technique requires a comparatively longer reaction time, higher temperature, and higher pressure. It also produces environmentally corrosive waste materials, which significantly increase the reaction costs due to handling and disposal requirements.
3.2. Base-Catalyzed Depolymerization of Lignin
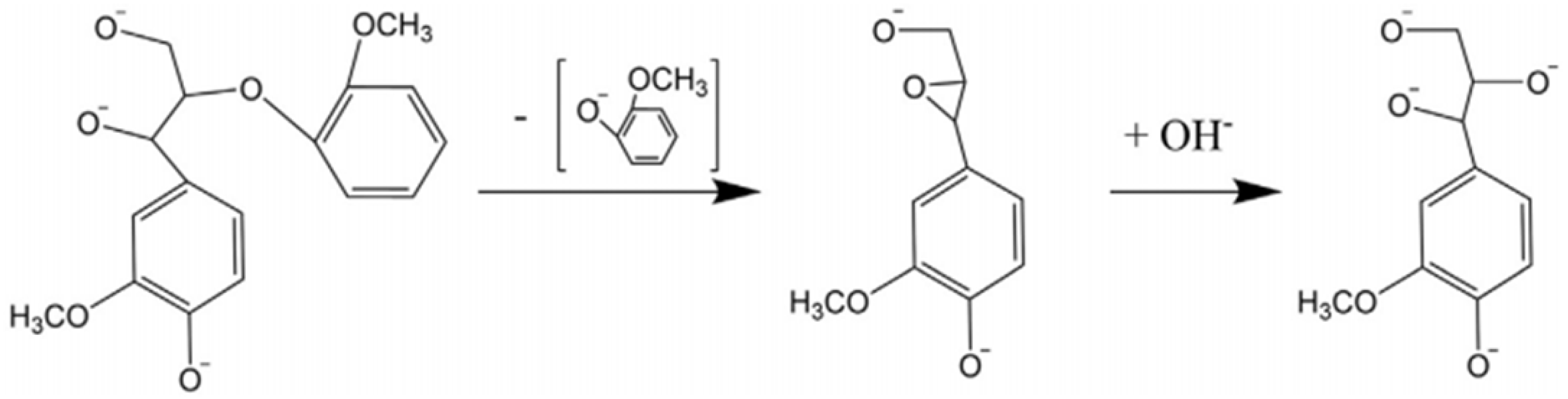
Base-catalyzed depolymerization (BCD) is another commonly used method to extract phenolic monomers from lignin. This method is performed at a higher temperature, where different bases, such as NaOH, KOH, and Ca(OH)
2, are used as catalysts [
68,
69]. This depolymerization technique is considered one of the most promising methods due to its excellent catalytic performance [
54,
70]. It is usually conducted at >300 °C and high pressure. The most abundant linkage in lignin is the β-O-4 bond; cleavage of this bond begins at 270 °C. During the reaction, cations from the base assist in forming cation adducts, which catalyze a six-membered transition on the β-O-4 bond resulting in the formation of phenolic monomers [
71]. A base catalyst mechanism is shown in
Figure 4.
BCD is also well known for its controlled hydrolytic cleavage of the ether bonds of lignin [
72]. A study has shown that weak bases, such as Ca(OH)
2 and LiOH, can produce smaller amounts of depolymerized product with lower depolymerization rates than strong bases, such as KOH and NaOH [
69]. Although the strong bases showed efficient depolymerization of lignin, bases such as LiOH, KOH, and CsOH were inefficient because of char deposition [
70]. BCD could be used to achieve high yields for selective monomeric products. Therefore, selectivity and yield depend on the temperature, pressure, reaction time, base concentration, and solvent type [
66,
73].
An alkaline-based catalyst is a homogeneous catalyst that causes complex post-separation. MgO is an inexpensive solid base catalyst alternative to the homogeneous catalyst. It is also considered robust, with excellent catalytic activity [
74]. The reaction conditions and promising outcomes using BCD of lignin are summarized in
Table 3.
Table 3 shows that most of the base-catalyzed transformations of lignin are performed at ≥300 °C and >10 MPa, where monomeric products vary with the reaction conditions. Beauchet et al. depolymerized KL at 270–315 °C and 130 bar at two different NaOH concentrations [
72]. Their results demonstrated that NaOH efficiently acted as a catalyst to increase the production of aromatic monomers. The monomeric production increased significantly with increased temperature, where the total yield of 8.4 wt% was reached at 315 °C. Pyrocatechol phenolic monomers were abundant (up to 25.8%) at 315 °C with high selectivity.
Besides temperature, pressure is another critical factor in obtaining a better yield. Lavoie et al. studied softwood lignin depolymerization using 5 wt% NaOH at temperatures of 300–330 °C and pressures of 9–13 MPa. Their chromatographic study identified 26 compounds; the major products were vanillin, guaiacol, and catechol [
70]. A similar study was performed by Roberts et al. at elevated pressure (25 MPa), and the major products, syringol, hydroxyacetophenone, and catechol, differed [
71]. These studies revealed that the nature of phenolic monomers depends significantly on the reaction pressure.
Solvents also play an important role in the BCD of lignin. Therefore, solvent selection is crucial for these reactions. A depolymerization reaction with a low base catalyst concentration in water shows very little depolymerization, sometimes yielding almost no depolymerized products [
83]. This indicates that an aqueous environment is not the correct solvent for this reaction. As a result, most researchers have used organic solvents, such as ethanol, isopropanol, tetrahydrofuran, and polyethylene glycerol, for BCD of lignin [
84,
85]. Long et al. have shown that tetrahydrofuran (THF) works well as a solvent for the depolymerization of lignin [
86]. Their study revealed that THF significantly promoted the catalytic activity of MgO. Hence, the production of phenolic monomers at 250 °C for 15 min increased by 13.2% compared to conventional solvents.
3.3. Oxidative Depolymerization of Lignin
Studies have shown that acid- or base-catalyzed depolymerization is a promising method to produce phenolic monomers by cleaving the ether linkages of lignin. However, these methods require relatively extreme reaction conditions, making the methods expensive, environmentally unfriendly, and difficult to handle.
Oxidative depolymerization is a promising method for converting lignin into monomers at milder conditions than the acid or base catalyst methods [
87]. A wide range of oxidants is used for the depolymerization reaction, such as permanganate, nitrobenzene, copper oxide, hydrogen peroxide, chlorine, and hypochlorite [
17,
88,
89]. The oxidative degradation approach selectively breaks down the ether linkages by preserving the aromatic character [
90]. Strong oxidants can break the linkages among the aromatic moieties of the lignin with good efficiency at milder conditions than catalysis methods [
8]. Therefore, successful oxidative depolymerization depends on the proper selection of the oxidant. Reaction conditions and the major outcomes of oxidative depolymerization of lignin are listed in
Table 4.
Hydrogen peroxide, an important oxidant, has been used to depolymerize lignin for the past few decades. It is an eco-friendly and economic catalyst that donates oxygen during lignin depolymerization [
98]. Studies have demonstrated that a minimum of 0.1% hydrogen peroxide can depolymerize the alkali lignin into monomers [
99]. Other studies have stated that H
2O
2 can selectively break down the β-O-4 and β-1 linkages at low temperatures [
100]. Although H
2O
2 shows a higher degradation rate than other methods, it produces a lower monophenolic compound yield. H
2O
2 in the presence of a metallic catalyst CuO (as Cu
2+) can cleave more ether bonds and side chains. Furthermore, in the presence of Fe
2(SO
4)
3 (as Fe
3+), the oxidation ability of H
2O
2 is significantly increased, which could increase the production of monophenols. In addition, the performance of hydrogen peroxide-based depolymerization can be significantly increased by acid [
100,
101].
Oxidative depolymerization can be performed without using metallic or other toxic materials. Wet air and molecular oxygen are considered less hazardous and more benign oxidants than metallic or traditional oxidants. Results have shown that wet air oxidation of lignin can substantially increase the production of phenolic monomers at low temperatures and pressures [
97]. Molecular oxygen depolymerization efficiently converts lignin into aromatic compounds in moderate reaction environments [
93].
3.4. Depolymerization of Lignin by Pyrolysis
Pyrolysis is another method frequently used to depolymerize lignin into its monomeric products. This method uses heat to convert the non-volatile compounds to a volatile mixture in the absence of oxygen. The pyrolytic mixture contains different aromatic monomer types, which indicates that the pyrolysis method could effectively convert lignin into biomaterials [
102]. It is a very fast method, taking only a few milliseconds to reach the pyrolytic temperature, compared to other thermal methods, for example, a thermogravimetric analyzer coupled with a Fourier Transform Infrared spectrometer (TG-FTIR) [
103]. The efficiency of the depolymerization and the molecular weight of the monomeric products can be modified by tuning the reaction conditions, such as the reaction time [
104], catalysts [
105], and the sources of lignin [
106]. The pyrolytic depolymerization of lignin starts with the cleavage of weak linkages at lower temperatures (<450 °C), followed by further breaking stronger linkages at higher temperatures (>450 °C). At higher temperatures, larger molecules decompose at selective sites to form smaller molecules, which are then separated by gas chromatography and detected by mass spectrometry [
107]. This method can be performed differently, but most pyrolysis methods are divided into two stages [
108]. The reaction conditions and promising outcomes of pyrolysis depolymerization of lignin are summarized in
Table 5.
The first pyrolysis stage occurs in the temperature range of 150–400 °C. Only basic side chains are broken during this stage, and other linkages, such as aromatic methylated groups and condensed linkages, remain stable [
106]. Therefore, softwood lignin produces a larger amount of residue than hardwood lignin because it contains more condensed and phenolic linkages; the major products in this stage are vanillin, coniferyl alcohol, isoeugenol, and other unsaturated compounds [
19,
117].
The second pyrolysis stage occurs in the temperature range between 400 °C and 800 °C. Most condensed methoxy groups and phenolic ether linkages are cleaved during this stage and the hydroxyl or methylated groups bond to the aromatic groups [
118]. Guaiacol,
o-vanillin, and
o-quinone are produced in this stage [
119]. The cleavage of benzene rings produces non-condensable gases at 550 °C [
120].
Pyrolysis depolymerization has been performed with different combinations of catalysts and solvents to obtain better performance. Since pyrolysis is performed in the absence of oxygen, catalysts and solvents may improve the performance of depolymerization by assisting in demethylation and preventing repolymerization and condensation by providing an oxidant or hydrogen donor. Studies showed that zeolite (ZSM-5) metal is a common catalyst and has two roles (acidic sites and pores), which help to control the reaction and yield more stable and desired products [
45]. Another study has proven that the zeolite (ZSM-5), along with a mixture of Si/Al, improved the pyrolysis efficiency and increased the production of aromatic monomers [
113,
117]. However, the phenolic character of lignin can also deactivate the zeolite [
121]. Some other studies have stated that Cu and Ni are better catalysts than zeolite and can selectively cleave the linkages in lignin to generate particular aromatic products [
122].
3.5. Microwave-Assisted Depolymerization of Lignin
Microwave-assisted technology is attractive because of its unique heating potential [
118]. This is an alternative fast method for degrading samples compared to traditional heating technology. Microwave electromagnetic radiation is applied to degrade the lignin into the value-added products [
123]. This method does not require physical contact between the heat source and the materials. It can create a huge amount of heat by rotating the polar molecules and causing ionic conduction [
21,
124]. Microwave-assisted technology is an economical and quick method for converting biomass and lignin into valuable products [
125]. The reaction conditions and promising outcomes of microwave-assisted depolymerization of lignin are summarized in
Table 6.
Recent studies have demonstrated that microwave-assisted technology has many advantages over traditional technology. It is fast, highly efficient, uniform, selective, and eco-friendly [
136]. The heating system for this method is favorable for large-scale industries because of the instantaneous start and stop ability. Research has shown that polar materials preferentially absorb more energy, which helps penetrate the interior part of the raw materials, significantly reduces the processing time, and eventually increases the reaction rate [
137]. This technology can add non-thermal effects to the reactions, which causes the reaction to proceed more quickly [
138]. Godey et al. have shown that microwave-assisted reactions significantly increased the reaction rates compared to conventional methods [
139].
Microwave-assisted technology can improve the reaction selectivity by cleaving the condensed bonds of lignin in the presence of a metal catalyst at lower temperatures. Zhu et al. used this method to depolymerize lignin into phenolic monomers through selective cleavage of the Cα-Cβ linkages [
140], using ferric sulfate as the catalyst. Their study showed that the microwave method could selectively break down the Cα-Cβ bonds in phenolic and non-phenolic dimers, where the cleavage rate for the phenolic dimer was significantly higher than for non-phenolic dimers. Furthermore, the metal catalyst significantly decreased the activation energy of the depolymerization reaction and inhibited the formation of char [
141].
Microwave-assisted technology could also play an important role in the liquefaction or solvolysis of lignin due to its rapid heating rate and high efficiency. Lignin can easily be dissolved in an IL at a lower temperature and for short periods using this method [
142]. Additionally, 1-butyl-3-methylimidazolium hydrogen sulfate (an IL) not only performs as an efficient catalyst for lignin depolymerization, but can also be recycled five times without loss of catalytic activity [
143].
3.6. Depolymerization of Lignin Using Sub- and Supercritical Fluids
Developing an alternative green technology with minimal environmental impact is important to produce specific desired products from lignin. Therefore, the reduction of energy consumption, the use of less toxic materials, and the efficient conversion of lignin are crucial requirements [
144,
145]. Supercritical solvent-based techniques have been considered an efficient, greener technology to depolymerize lignin to successfully prevent condensation reactions [
146,
147]. Sub- and supercritical fluids are formed under extremely high temperatures and pressures. Supercritical fluids exhibit distinct properties, such as low viscosity, low dielectric constant, and high diffusivity, which penetrate the structure of the lignin and easily solubilize the depolymerized products [
148].
Supercritical methods have been used since the 1990s, and during the initial stage of their use, KL and OL were treated with supercritical methanol [
73]. Different types of sub- and supercritical fluids, such as water [
149], ethanol [
150], and carbon dioxide [
151], have been used. Condensation is very common in depolymerization, but supercritical depolymerization reactions are highly efficient at preventing condensation. Studies have shown that intermediates formed from the lignin can couple with the hydrogen radicals generated from the supercritical fluids and prevent the condensation reaction [
51,
60]. The reaction conditions of sub- and supercritical depolymerization of lignin are summarized in
Table 7.
Wahyudiono et al. successfully conducted supercritical water depolymerization of lignin at 350–400 °C and 25–40 MPa [
160]. They showed that the formation of heavier compounds increased with temperature, and the formation of heavier compounds decreased with reaction time. Catechol,
m,
p-cresol,
o-cresol, and phenol were identified as the major products, which indicated that water is an outstanding solvent for converting lignin into monomers. Another study was performed by Numan-Al-Mobin et al. on the depolymerization of lignin with a mixture of water and subcritical CO
2 (ScCO
2) [
151]. Subcritical CO
2 is a naturally available compound known for its catalytic properties, sustainability, and non-flammable nature. The combined use of water and subcritical CO
2 was an efficient, greener method for synthesizing phenolic monomers.
Depolymerization of lignin has also been performed using sub- and supercritical ethanol, methanol, and combined with water. This study showed that ethanol can significantly enhance the depolymerization of lignin and decrease char formation [
150]. This result may be due to the formation of hydrogen radicals from the ethanol, which is coupled with the intermediates from lignin and inhibits the repolymerization reaction intermediates from forming char. In contrast, supercritical methanol has some interesting properties (e.g., hydrogen donation ability, high heat transfer, high dispersity, and improvement of catalytic activity), which favor the depolymerization of lignin [
157]. The depolymerization of lignin into phenolic compounds can also be performed by mixing sub- and supercritical ethanol and water [
152]. Supercritical ethanol is more effective than methanol at liquefying the woody biomass into crude products. Additionally, supercritical ethanol is expected to dissolve and stabilize the liquid products more easily. The following schematic diagram summarizes different depolymerization methods (
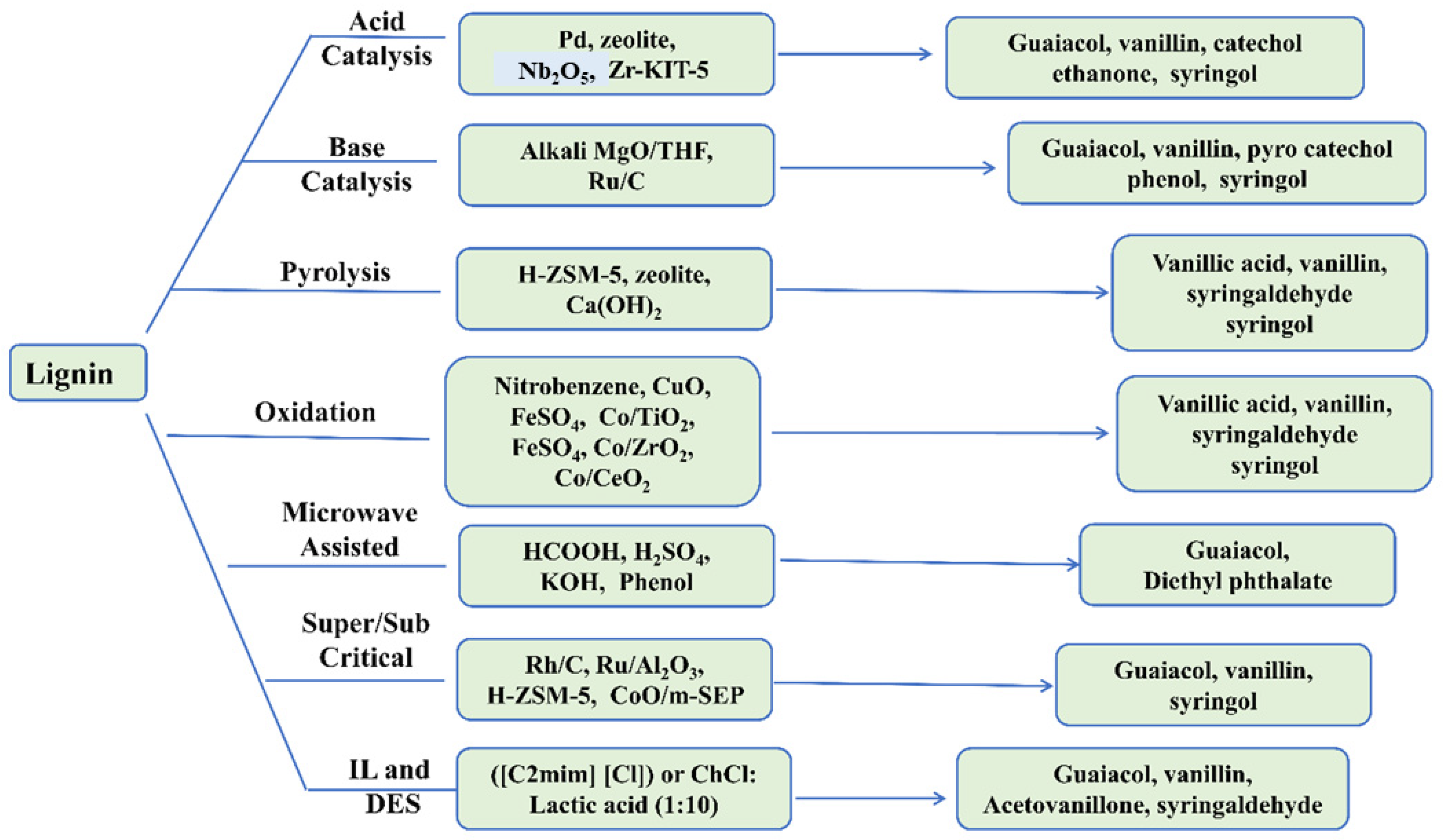
Figure 5).
3.7. Depolymerization of Lignin by Ionic Liquids and Deep Eutectic Solvents
The previously discussed methods are very effective for depolymerizing lignin into aromatic compounds. However, using corrosive chemicals, extreme reaction conditions, and harmful waste production increases the processing cost. Therefore, a sustainable green method is needed to depolymerize lignin into aromatic monomers to reduce production waste, avoid harming the environment, and decrease processing costs. Currently, ILs are considered a green technology for depolymerizing lignin through the cleavage of β-O-4 bonds. ILs show numerous characteristics desirable for a green medium, such as low cost of formulation, non-volatile, chemical inertness, low viscosity, acidity, and excellent miscibility [
161,
162]. In addition, since they are liquid over a wide range of temperatures and show thermal, chemical, and electrochemical stability, ILs have numerous applications and are suitable environmentally friendly alternative solvents for the depolymerization of lignin [
163].
Recent studies have reported that ILs are an excellent solvent for fractionating lignocellulose into its basic components and the solubilization of lignin, since they are long-range, noncovalent, interaction-based liquid formulations. The interactions among their cations and anions help break the bonds of lignin and help them to keep their state in a liquid phase, typically below 100 °C [
162,
164,
165]. ILs can potentially control the degree of oxidative depolymerization and cooperate with different catalysts [
166]. Consequently, ILs have gained much attention from the scientific community as a method of depolymerizing lignin into value-added products. Numerous combinations of ionic liquids with metal catalysts, such as Co, Cu, and Mn, have been studied to depolymerize lignin. Stark et al. have converted lignin with combinations of different ionic liquids and the catalyst Mn(NO
3)
2. They have reported that the combination of Mn(NO
3)
2 and 1-ethyl-3-methylimidazolium trifluoromethanesulfonate [EMIM][CF
3SO
3] was the most effective reaction medium for lignin depolymerization. They also found that more than 63% of organosolv beach lignin can be selectively converted into phenolic monomers with 11.5 wt% of 2,6-dimethoxy-1,4-benzoquinone at 100 °C for 24 h [
87]. Li et al. converted organosolv bagasse lignin into phenolic monomers using cooperative [bmim][CF
3SO
3]/[bSmim][HSO
4] and water [
167]. They reported that the conversion of lignin reached 66.7% with 14.5 wt% of phenolic compounds and negligible char formation at milder reaction conditions (250 °C, 30 min). At 250 °C, water is subcritical, which converted 36.4% of the lignin into phenolic monomers by self-catalytic activity due to the improved dissociation of H
+. Most lignin is converted to unwanted char product at this temperature due to the carbocation mechanism [
168]. Nevertheless, [bmim][CF
3SO
3] is an excellent hydrogen bond donor, where the conversion of lignin increased to 45.2% without any char formation by facilitating the cleavage of the hydrogen-bonded linkages [
169]. After the addition of an acidic IL [bSmim][HSO
4], the conversion of lignin reached 60.1%, and when the concentration of IL [bSmim][HSO
4] increased from 2.0 to 3.0 mmol, the conversion reached 66.7%.
ILs can act as acids, bases, and nucleophiles because of their high structural flexibility. Therefore, ILs can mimic the acid- or base-catalyzed depolymerization of lignin without using any corrosive chemicals. The efficient depolymerization of lignin by ILs depends significantly on the cations and anions of the ILs [
170]. Recent studies have shown that the anions primarily affect the integrity of the lignin structure, while cations act like spectators [
171]. Another study showed that this anionic activity is affected by the strength of the corresponding coordination interaction with the hydrogen in the hydroxyl group of the lignin backbone and the lignin-like structures [
172].
Figure 6 shows that the coordination with hydrogen directs the nucleophilic attack toward the carbon double bond by stabilizing the electronic environment of the compounds. Tolesa et al. used an ammonium-based IL to selectively depolymerize the lignin into phenolic monomers. Two different types of ionic liquids, equimolar diisopropylethylamine (DIPEA) and acetic acid (A) or octanoic acid (O), were used as a solvent for the selective depolymerization of lignin. They have shown that ILs not only act as a solvent, but also as a catalyst [
173]. Cox et al. investigated how the anion of an acidic IL can stabilize the hydroxyl groups of lignin compounds, resulting in more rapid cleavage of the C–O bonds of lignin [
174].
DES-based depolymerization is one of the newest approaches in lignin bond depolymerization. Electrochemical catalysis, microwave-irradiated, and typical heating methods using deep eutectic solvents are the most common methods used so far [
133,
175,
176,
177]. The efficacy of these methods compared to the conventional methods has been characterized and monitored using
13C-NMR,
1H-NMR, two-dimensional-heteronuclear single quantum correlation (2D-HSQC) NMR,
31P NMR, cyclic voltammetry (CV), gel permeation chromatography (GPC), scanning electron microscopy (SEM) techniques, and gas chromatography−mass spectrometry (GC−MS) [
133,
175,
176,
177]. The following methods and their significant outcomes are tabulated in
Table 8.
The most common solvent used to dissolve KL is a NaOH solution. This basic solution is suitable for the electrochemical reaction due to its high conductivity. Typical interlinkages, such as C–C and C–O bonds in lignin, can be cleaved by catalytic properties using an appropriate electrode potential [
177]. The major shortcomings of this process are selectivity, low yields at mild conditions, and consecutive oxidations of the products producing unwanted CO
2 and organic acids. The high pH also limits the choice of electrode materials to a few metals [
177].
After the DES-based depolymerization, lignin recovery is also an important step. Recovery can be made by diluting the reaction mixture with a diluted (0.01 M) H
2SO
4 solution, followed by the precipitation of insoluble lignin [
177]. Then, the solution is further dissolved in 1 M NaOH for size-exclusion chromatography analysis. The soluble part of lignin is extracted from the supernatant using the liquid–liquid extraction (LLE) technique, where ethyl acetate is preferable as an organic solvent because it is greener. The aromatic compounds produced in the depolymerization step can be extracted with methyl isobutyl ketone (MIBK) and the LLE technique for further analysis. Both EA and MIBK phases should be well-mixed for a few hours [
177].
In the case of alkali lignin depolymerization, ChCl (choline chloride), more specifically chloride ions, works as a bridge to form hydrogen bonds with the -OH groups of lignin and methanol [
23,
182]. This mechanism could be responsible for the enhanced dissolution and mass transfer efficiency of the lignin depolymerization reaction [
175].
Two-dimension-HSQC NMR spectra analysis indicates that the addition of water can break the interactions among hydrogen bond acceptors (HBAs) and hydrogen bond donors (HBDs) as a deep eutectic solvent (DES) is formulated by mixing HBA and HBD molecules under simple heating and stirring approaches [
175]. Thus, the DES system decreases the solubility of lignin, and condensation and depolymerization of the side chains of the alkali lignin can occur simultaneously [
175]. If the side chains are longer, they might inhibit acetic acid formation, and higher amounts of guaiacyl moieties might increase the yield of acetovanillone. DES could enhance the catalytic activities of metal-based catalysts by improving their redox potential [
175].
The selective cleavage of H-moieties can be achieved using a metal-based DES, such as 1:2 ChCl:FeCl
3. This DES can selectively cleave the ester or β-O-4 bonds of the lignin structure, which eventually produces p-coumaric acid (pCA) units [
181]. Careful modification of pCA produces desirable methyl p-hydroxycinnamate (MPC). The metal-based DES catalyst is recyclable, easy to prepare, and economical. This method is even effective for corncob lignin and can produce 116.6 mg/g of MPC [
181].
The efficacy of depolymerization techniques can be further improved by applying microwave heating, which significantly reduces the reaction time [
133]. NMR, GPC, and molecular dynamics simulations show that microwave heating selectively cleaves the target C–C and ether bonds, providing a narrower MW distribution [
133]. Microwave irradiation stretches specific lignin bonds, increasing the probability of the bond breaking.
Overall, it has been shown that DES-based lignin depolymerization provides more selectivity, higher yield, uses greener and recyclable chemicals, and requires lower temperatures and less time. Comparative advantages and disadvantages of different greener depolymerization techniques has been summarized in the following table (
Table 9).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}