
Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 3: Mature Leukemias/Lymphomas


Abstract
:1. Introduction
2. An Overview of Incidence, Mortality, and Survival Data for the Types of Lymphoid Neoplasms
3. Mature B-Cell Neoplasms
3.1. Lymphomas in the Pediatric and Adult Age Groups
3.2. Bruton’s Tyrosine Kinase Inhibitors as an Example of Precision Medicine
3.3. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
- (1)
- SETD2/del3p21.31, del9p21.3, and gains of chr17q21.31 are associated with relapsed/refractory disease and TP53 disruption.
- (2)
- MED12 and DDX3X mutations are associated with unmutated IGHV CLL.
- (3)
- B-cell receptor immunoglobulin subset 2, which represents about 3% of all CLL and is known to be associated with a poor prognosis, is linked to the putative driver, FAM50A.
- (4)
- IGHV3-21 gene rearrangement is enriched for FAM50A, ATM/del11q22, SF3B1 mutations, and chr21q21.3-q22.3 gains.
3.3.1. CLL Diagnosis and Prognosis
3.3.2. Other Relevant Diagnostic Aspects in B-Cell Neoplasms in Relation to CLL in WHO-HAEM5
WHO-HAEM5 Eliminated B-Prolymphocytic Leukemia
Splenic B-Cell Lymphomas/Leukemias in WHO-HAEM5
- (1)
- Small B-cell lymphoma involving bone marrow, peripheral blood, or both, composed of small lymphoid cells with villous processes;
- (2)
- Neoplastic cells express pan-B-cell markers, IgM, and IgD and are negative for BCL6, annexin A1, CD103, cyclin D1, SOX11, and LEF1;
- (3)
- Other splenic and nodal B-cell lymphomas should be excluded; and
- (4)
- Clinical or imaging studies should show splenomegaly.
3.4. Follicular Lymphoma
3.4.1. Grading in Follicular Lymphoma
3.4.2. Other Diagnostic Considerations in Follicular Lymphoma
- (1)
- Intermediate or large-cell morphology;
- (2)
- Follicular, diffuse, or follicular and diffuse growth pattern;
- (3)
- Mature B-cell phenotype with co-expression of B-cell lymphoma 6 (BCL6) and MUM1 proteins, and
- (4)
- IRF4 translocation.
3.5. In Situ Mantle Cell Neoplasia in Relation to Overt Mantle Cell Lymphoma
3.6. Lymphoplasmacytic Lymphoma
3.7. Immunophenotypic Features Characteristic for Specific Mature B-Cell Lymphomas/Leukemias Composed of Small to Medium-Sized Neoplastic Cells
3.8. Aggressive Mature B-Cell Lymphomas
3.8.1. Burkitt Lymphoma
3.8.2. How the Classification of Aggressive Non-Burkitt (or large) B-Cell Lymphomas Has Evolved
3.8.3. DLBCL/High-Grade B-Cell Lymphoma with MYC and BCl2 Rearrangements
3.8.4. High-Grade B-Cell Lymphoma with Chromosome 11q Aberration
3.8.5. Primary Large B-Cell Lymphoma of Immune-Privileged Sites
3.8.6. Intravascular Large B-Cell Lymphoma
3.9. Immunophenotypic Features Characteristic of Mature B-Cell Lymphomas/Leukemias Composed of Medium-Sized to Large Cells
3.10. Primary Mediastinal Lymphomas
Mediastinal Grey Zone Lymphoma: In the Middle of the Spectrum between Classic Hodgkin Lymphoma and Primary Mediastinal Large B-CELL lymphoma
- (1)
- A confluent growth of pleomorphic cells within a variably abundant microenvironment and dense fibrotic stroma;
- (2)
- Uniform strong expression of CD20 and uniform strong PAX5 and uniform and strong expression of at least one additional B-cell marker, CD19, CD79a, BOB1, and OCT2;
- (3)
- CD30 positive expression, with varying intensity.
3.11. Immunohistochemical Features of Hodgkin Lymphomas, Peripheral T-Cell Lymphomas, and Other Lymphomas That May Be in the Differential Diagnosis
4. Mature T-Cell and NK-Cell Neoplasms
4.1. Name Changes in WHO-HAEM5 and ICC Compared with the WHO 2017 Classification
4.2. Nodal T-Follicular Helper (TFH) Cell Lymphoma
- (1)
- Nodal disease,
- (2)
- CD4+, occasionally CD4-negative, CD8-negative atypical lymphoid cells,
- (3)
- Extrafollicular FDC expansion, and
- (4)
- HEV hyperplasia, which is mild in tumor-cell-rich cases.
4.3. Anaplastic Large-Cell Lymphoma
- (1)
- Complete or partial infiltration of lymph node or extranodal tissue by large pleomorphic cells with lobated nuclei, distinct nucleoli, including “hallmark cells,”
- (2)
- Uniform strong expression of CD30,
- (3)
- Absence of ALK protein expression or ALK rearrangement, and
- (4)
- Negativity for EBV. The desirable criteria include expression of T-cell markers and cytotoxic markers, albeit with frequent losses, and clonal rearrangement of the TCR gene [12].
4.4. EBV+ Nodal T- and NK-Cell Lymphoma
4.5. Peripheral T-Cell Lymphoma, Not Otherwise Specified (PTCL NOS)
- (1)
- The presence of an abnormal T-cell infiltrate, which is morphologically or immunophenotypically aberrant, monoclonal by ancillary studies, or both;
- (2)
- The tumor cells are negative or express only one TFH marker (to differentiate from nodal TFH cell lymphomas), and the neoplasm only shows EBER positivity in scattered B-cells (to differentiate from EBV+ nodal T- and NK-cell lymphoma);
- (3)
- The exclusion of other nodal or extranodal mature T- and NK-cell lymphomas, i.e., ALK+ ALCL, ALK-negative ALCL, adult T-cell leukemia/lymphoma, extranodal NK/T-cell lymphoma.
5. Genetic Predisposition and Constitutional Inherited Syndromes
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | activated B-cell |
| AESOP | adenopathy and extensive skin patch overlying a plasmacytoma |
| AID | activation-induced cytidine deaminase |
| AIDS | acquired immune deficiency syndrome |
| AITL | angioimmunoblastic T-cell lymphoma |
| ALCL | anaplastic large-cell lymphoma |
| ALK | anaplastic lymphoma kinase |
| ALL | acute lymphoblastic leukemia |
| ASR | age-standardized rate |
| AYA | adolescents and young adults |
| BL | Burkitt lymphoma |
| B-PLL | B-cell prolymphocytic leukemia |
| BTK | Bruton’s tyrosine kinase |
| CDR3 | complementarity-determining region 3 |
| CHL | classic Hodgkin lymphoma |
| CHOP | cyclophosphamide, doxorubicin, vincristine, and prednisolone |
| CLL | chronic lymphoid leukemia |
| CNS | central nervous system |
| DLBCL | diffuse large B cell lymphoma |
| DLBCL NOS | diffuse large B-cell lymphoma, not otherwise specified |
| EBV | Epstein–Barr virus |
| FCI | flow cytometric immunophenotyping |
| FDA | Food and Drug Administration |
| FDC | Follicular dendritic cells |
| FISH | Fluorescence in situ hybridization |
| FL | follicular lymphoma |
| FOXO1 | forkhead box O1 |
| GC | germinal center |
| GCB | germinal center B-cell |
| GI | gastrointestinal |
| GLOBOCAN | Global Cancer Observatory |
| GNA13 | G protein subunit alpha 13 |
| H&E | Hematoxylin and eosin |
| HCL | hairy cell leukemia |
| HEVs | high endothelial venules |
| HGBCL | high grade B-cell lymphoma |
| HGBCL-11q | high-grade B-cell lymphoma with chromosome 11q aberration |
| HL | Hodgkin lymphoma |
| HMRN | Hematologic Malignancy Research Network |
| HRS | Hodgkin or Reed–Sternberg |
| ICC | International Consensus Classification |
| IDD | immune deficiency or dysregulation |
| IHC | immunohistochemical |
| ILL | intermediate lymphocytic lymphoma |
| IRAK | IL-1R-associated kinase |
| ISMCN | in situ mantle cell neoplasia (or in situ mantle cell neoplasm) |
| IVBCL | intravascular large B-cell lymphoma |
| L&H | lymphohistiocytic |
| LBCL-IRF4 | large B-cell lymphoma with an IRF4 rearrangement |
| LMO2 | LIM-domain only 2 |
| LMP1 | latent membrane protein 1 |
| LOH | loss of heterozygosity |
| LPD | lymphoproliferative disorder |
| LPL | lymphoplasmacytic lymphoma |
| MALT | mucosa-associated lymphoid tissue |
| MBL | monoclonal B-cell lymphocytosis |
| MCL | mantle cell lymphoma |
| MGUS | monoclonal gammopathy of undetermined significance |
| MGZL | mediastinal gray (or grey) zone lymphoma |
| MZ | mantle zone |
| MZL | marginal zone lymphoma |
| NGS | next-generation sequencing |
| NFκB | nuclear factor kappa B |
| NHL | non-Hodgkin lymphoma |
| NK | Natural Killer |
| NLPHL | nodular lymphocyte predominant Hodgkin lymphoma |
| NOS | not otherwise specified |
| PCNS-LBCL | Primary large B-cell lymphoma of the CNS |
| PMBCL | primary mediastinal large B-cell lymphoma |
| POEMS | polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes |
| PTCL | peripheral T-cell lymphoma |
| PTCL NOS | peripheral T-cell lymphoma, not otherwise specified |
| PTFL | pediatric-type follicular lymphoma |
| PTLD | post-transplant lymphoproliferative disorder |
| PVR-LBCL | primary vitreoretinal large B-cell lymphoma |
| REAL | Revised European American Lymphoma Classification |
| SEER | Surveillance, Epidemiology, and End Results |
| SLL | small lymphocytic lymphoma |
| SMZL | splenic marginal zone lymphoma |
| SOX11 | sex-determining region Y-box transcription factor 11 |
| TEMPI | telangiectasias, erythrocytosis with elevated erythropoietin, monoclonal gammopathy, perinephric fluid collections, intrapulmonary shunting |
| TFH | T-follicular helper |
| WHO | World Health Organization |
| WHO-HAEM5 | Fifth edition of WHO classification for hematolymphoid tumors |
| WM | Waldenström macroglobulinemia |
References
- The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997, 89, 3909–3918. [Google Scholar] [CrossRef]
- Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 1: Lymphoid Neoplasms. Lymphatics 2023, 1, 55–76. [Google Scholar] [CrossRef]
- Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 2: B-/T-Cell Acute Lymphoblastic Leukemias. Lymphatics 2023, 1, 118–154. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Blum, K.A.; Keller, F.G.; Castellino, S.; Phan, A.; Flowers, C.R. Incidence and outcomes of lymphoid malignancies in adolescent and young adult patients in the United States. Br. J. Haematol. 2018, 183, 385–399. [Google Scholar] [CrossRef] [Green Version]
- The United States National Cancer Institute Surveillance, Epidemiology, and End Results Program Database. Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/ (accessed on 25 June 2023).
- Al-Hamadani, M.; Habermann, T.M.; Cerhan, J.R.; Macon, W.R.; Maurer, M.J.; Go, R.S. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am. J. Hematol. 2015, 90, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef]
- The United States National Cancer Institute Surveillance, Epidemiology, and End Results Program Database. Browse the SEER Cancer Statistics Review (CSR) 1975–2011. Table 19.29. All Lymphoid Neoplasms with Detailed Non-Hodgkin Lymphoma Subtypes. Available online: https://seer.cancer.gov/archive/csr/1975_2011/browse_csr.php?sectionSEL=19&pageSEL=sect_19_table.29.html (accessed on 23 June 2023).
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Hematolymphoid Tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2022. (WHO Classification of Tumours Series, 5th ed.; vol. 11). Available online: https://https://tumourclassification.iarc.who.int/home (accessed on 16 May 2023).
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- SEER*Explorer: An Interactive Website for SEER Cancer Statistics [Internet]. Surveillance Research Program, National Cancer Institute; 19 April 2023. Data Source(s): SEER Incidence Data, November 2022 Submission (1975–2020), SEER 22 Registries. Available online: https://seer.cancer.gov/statistics-network/explorer/ (accessed on 14 May 2023).
- American Cancer Society. About Non-Hodgkin Lymphoma in Children. Available online: https://www.cancer.org/cancer/types/childhood-non-hodgkin-lymphoma/about.html (accessed on 14 May 2023).
- National Cancer Institute. Cancer in Children and Adolescents. Available online: https://www.cancer.gov/types/childhood-cancers/child-adolescent-cancers-fact-sheet#r2 (accessed on 14 May 2023).
- Kansal, R. Germline predisposition in hematologic malignancies. In Comprehensive Hematology and Stem Cell Research; Rezaei, N., Ed.; Elsevier: Amsterdam, The Netherlands, 2023; submitted. [Google Scholar]
- Minard-Colin, V.; Brugières, L.; Reiter, A.; Cairo, M.S.; Gross, T.G.; Woessmann, W.; Burkhardt, B.; Sandlund, J.T.; Williams, D.; Pillon, M.; et al. Non-Hodgkin Lymphoma in Children and Adolescents: Progress Through Effective Collaboration, Current Knowledge, and Challenges Ahead. J. Clin. Oncol. 2015, 33, 2963–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandlund, J.T.; Martin, M.G. Non-Hodgkin lymphoma across the pediatric and adolescent and young adult age spectrum. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 589–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.J.; Długosz-Danecka, M.; Connors, J.M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol. 2021, 8, e410–e421. [Google Scholar] [CrossRef]
- Castellino, S.M.; Pei, Q.; Parsons, S.K.; Hodgson, D.; McCarten, K.; Horton, T.; Cho, S.; Wu, Y.; Punnett, A.; Dave, H.; et al. Brentuximab Vedotin with Chemotherapy in Pediatric High-Risk Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 1649–1660. [Google Scholar] [CrossRef]
- Conley, M.E.; Dobbs, A.K.; Farmer, D.M.; Kilic, S.; Paris, K.; Grigoriadou, S.; Coustan-Smith, E.; Howard, V.; Campana, D. Primary B cell immunodeficiencies: Comparisons and contrasts. Annu. Rev. Immunol. 2009, 27, 199–227. [Google Scholar] [CrossRef]
- Smith, C.I.; Baskin, B.; Humire-Greiff, P.; Zhou, J.N.; Olsson, P.G.; Maniar, H.S.; Kjellén, P.; Lambris, J.D.; Christensson, B.; Hammarström, L.; et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J. Immunol. 1994, 152, 557–565. [Google Scholar] [CrossRef]
- Neys, S.F.H.; Rip, J.; Hendriks, R.W.; Corneth, O.B.J. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 2021, 81, 1605–1626. [Google Scholar] [CrossRef]
- Baecklund, E.; Smedby, K.E.; Sutton, L.A.; Askling, J.; Rosenquist, R. Lymphoma development in patients with autoimmune and inflammatory disorders--what are the driving forces? Semin. Cancer Biol. 2014, 24, 61–70. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Shabani, M.; Tabary, M.; Rayzan, E.; Rezaei, N. Lymphoma in the setting of autoimmune diseases: A review of association and mechanisms. Crit. Rev. Oncol. Hematol. 2020, 150, 102945. [Google Scholar] [CrossRef]
- Grassilli, E.; Cerrito, M.G.; Lavitrano, M. BTK, the new kid on the (oncology) block? Front. Oncol. 2022, 12, 944538. [Google Scholar] [CrossRef] [PubMed]
- Imbruvica (Ibrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/205552s030,210563s006lbl.pdf (accessed on 25 March 2023).
- Calquence (Acalabrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/210259s009lbl.pdf (accessed on 25 March 2023).
- National Cancer Institute. Zanubrutinib’s Approval Improves Targeted Treatment for CLL. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2023/fda-zanubrutinib-cll-sll (accessed on 25 March 2023).
- Brukinsa (Zanubrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213217s005lbl.pdf (accessed on 25 March 2023).
- Food and Drug Administration. Drug Approvals and Databases. 27 January 2023. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pirtobrutinib-relapsed-or-refractory-mantle-cell-lymphoma (accessed on 25 March 2023).
- Rai, K.R.; Jain, P. Chronic lymphocytic leukemia (CLL)-Then and now. Am. J. Hematol. 2016, 91, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattone, L.; Ghia, P.; Scarfò, L. The evolving treatment landscape of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2019, 31, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Treatment of Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2020, 383, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Albiol, N.; Arguello-Tomas, M.; Moreno, C. The road to chemotherapy-free treatment in chronic lymphocytic leukaemia. Curr. Opin. Oncol. 2021, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.M.; Li, J.Y.; Gale, R.P.; Huang, X.J. The mystery of chronic lymphocytic leukemia (CLL): Why is it absent in Asians and what does this tell us about etiology, pathogenesis and biology? Blood Rev. 2015, 29, 205–213. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Landgren, O.; Albitar, M.; Ma, W.; Abbasi, F.; Hayes, R.B.; Ghia, P.; Marti, G.E.; Caporaso, N.E. B-cell clones as early markers for chronic lymphocytic leukemia. N. Engl. J. Med. 2009, 360, 659–667. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Green, M.J.; Kuzmicki, A.; Kennedy, B.; Fenton, J.A.; Evans, P.A.; O’Connor, S.J.; Richards, S.J.; Morgan, G.J.; Jack, A.S.; et al. Monoclonal B lymphocytes with the characteristics of “indolent” chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts. Blood 2002, 100, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Marti, G.E.; Rawstron, A.C.; Ghia, P.; Hillmen, P.; Houlston, R.S.; Kay, N.; Schleinitz, T.A.; Caporaso, N.; International Familial CLL Consortium. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br. J. Haematol. 2005, 130, 325–332. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Shanafelt, T.; Lanasa, M.C.; Landgren, O.; Hanson, C.; Orfao, A.; Hillmen, P.; Ghia, P. Different biology and clinical outcome according to the absolute numbers of clonal B-cells in monoclonal B-cell lymphocytosis (MBL). Cytom. B Clin. Cytom. 2010, 78 (Suppl. S1), S19–S23. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Kay, N.E.; Rabe, K.G.; Call, T.G.; Zent, C.S.; Maddocks, K.; Jenkins, G.; Jelinek, D.F.; Morice, W.G.; Boysen, J.; et al. Brief report: Natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 3959–3963. [Google Scholar] [CrossRef] [Green Version]
- Goldin, L.R.; Pfeiffer, R.M.; Li, X.; Hemminki, K. Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: Results from the Swedish Family-Cancer Database. Blood 2004, 104, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Slager, S.L.; Caporaso, N.E.; de Sanjose, S.; Goldin, L.R. Genetic susceptibility to chronic lymphocytic leukemia. Semin. Hematol. 2013, 50, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Kleinstern, G.; Weinberg, J.B.; Parikh, S.A.; Braggio, E.; Achenbach, S.J.; Robinson, D.P.; Norman, A.D.; Rabe, K.G.; Boddicker, N.J.; Vachon, C.M.; et al. Polygenic risk score and risk of monoclonal B-cell lymphocytosis in caucasians and risk of chronic lymphocytic leukemia (CLL) in African Americans. Leukemia 2022, 36, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Tiao, G.; Improgo, M.R.; Kasar, S.; Poh, W.; Kamburov, A.; Landau, D.A.; Tausch, E.; Taylor-Weiner, A.; Cibulskis, C.; Bahl, S.; et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017, 31, 2244–2247. [Google Scholar] [CrossRef] [Green Version]
- Lampson, B.L.; Gupta, A.; Tyekucheva, S.; Mashima, K.; Petráčková, A.; Wang, Z.; Wojciechowska, N.; Shaughnessy, C.J.; Baker, P.O.; Fernandes, S.M.; et al. Rare Germline ATM Variants Influence the Development of Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2023, 41, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): A meta-analysis of individual patient data. Lancet Oncol. 2016, 17, 779–790. [Google Scholar] [CrossRef]
- Seifert, M.; Sellmann, L.; Bloehdorn, J.; Wein, F.; Stilgenbauer, S.; Dürig, J.; Küppers, R. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J. Exp. Med. 2012, 209, 2183–2198. [Google Scholar] [CrossRef]
- Sutton, L.A.; Rosenquist, R. Deciphering the molecular landscape in chronic lymphocytic leukemia: Time frame of disease evolution. Haematologica 2015, 100, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Belessi, C.; Moreno, C.; Boudjograh, M.; Guida, G.; Smilevska, T.; Belhoul, L.; Stella, S.; Stavroyianni, N.; Crespo, M.; et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood 2007, 109, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2017, 31, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, P.M.; Hosnijeh, F.S.; Späth, F.; Hengeveld, P.J.; Agathangelidis, A.; Saleh, M.; Casabonne, D.; Benavente, Y.; Jerkeman, M.; Agudo, A.; et al. High-risk subtypes of chronic lymphocytic leukemia are detectable as early as 16 years prior to diagnosis. Blood 2022, 139, 1557–1563. [Google Scholar] [CrossRef]
- Kolijn, P.M.; Muggen, A.F.; Ljungström, V.; Agathangelidis, A.; Wolvers-Tettero, I.L.M.; Beverloo, H.B.; Pál, K.; Hengeveld, P.J.; Darzentas, N.; Hendriks, R.W.; et al. Consistent B Cell Receptor Immunoglobulin Features Between Siblings in Familial Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 11, 740083. [Google Scholar] [CrossRef]
- Davi, F.; Langerak, A.W.; de Septenville, A.L.; Kolijn, P.M.; Hengeveld, P.J.; Chatzidimitriou, A.; Bonfiglio, S.; Sutton, L.A.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene analysis in chronic lymphocytic leukemia in the era of next generation sequencing. Leukemia 2020, 34, 2545–2551. [Google Scholar] [CrossRef]
- Hengeveld, P.J.; Levin, M.D.; Kolijn, P.M.; Langerak, A.W. Reading the B-cell receptor immunome in chronic lymphocytic leukemia: Revelations and applications. Exp. Hematol. 2021, 93, 14–24. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbe, P.; Ridout, K.E.; Vavoulis, D.V.; Dréau, H.; Kinnersley, B.; Denny, N.; Chubb, D.; Appleby, N.; Cutts, A.; Cornish, A.J.; et al. Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features. Nat. Genet. 2022, 54, 1675–1689. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Rawstron, A.C.; Kreuzer, K.A.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Villamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom. B Clin. Cytom. 2018, 94, 121–128. [Google Scholar] [CrossRef]
- Giné, E.; Martinez, A.; Villamor, N.; López-Guillermo, A.; Camos, M.; Martinez, D.; Esteve, J.; Calvo, X.; Muntañola, A.; Abrisqueta, P.; et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica 2010, 95, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [Green Version]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Baliakas, P.; Espinet, B.; Mellink, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. Hemasphere 2022, 6, e707. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jäger, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agathangelidis, A.; Chatzidimitriou, A.; Chatzikonstantinou, T.; Tresoldi, C.; Davis, Z.; Giudicelli, V.; Kossida, S.; Belessi, C.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: The 2022 update of the recommendations by ERIC, the European Research Initiative on CLL. Leukemia 2022, 36, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Favini, C.; Talotta, D.; Almasri, M.; Andorno, A.; Rasi, S.; Adhinaveni, R.; Kogila, S.; Awikeh, B.; Schipani, M.; Boggione, P.; et al. Clonally unrelated Richter syndrome are truly de novo diffuse large B-cell lymphomas with a mutational profile reminiscent of clonally related Richter syndrome. Br. J. Haematol. 2022, 198, 1016–1022. [Google Scholar] [CrossRef]
- Siebert, R.; Schuh, A.; Ott, G.; Cree, I.A.; Du, M.Q.; Ferry, J.; Hochhaus, A.; Naresh, K.N.; Solary, E.; Khoury, J.D. Response to the Comments from the Groupe Francophone de Cytogénétique Hématologique (GFCH) on the 5th edition of the World Health Organization classification of haematolymphoid tumors. Leukemia 2023, 37, 1170–1172. [Google Scholar] [CrossRef]
- Chapiro, E.; Pramil, E.; Diop, M.; Roos-Weil, D.; Dillard, C.; Gabillaud, C.; Maloum, K.; Settegrana, C.; Baseggio, L.; Lesesve, J.F.; et al. Genetic characterization of B-cell prolymphocytic leukemia: A prognostic model involving MYC and TP53. Blood 2019, 134, 1821–1831. [Google Scholar] [CrossRef] [Green Version]
- Kansal, R.; Ross, C.W.; Singleton, T.P.; Finn, W.G.; Schnitzer, B. Histopathologic features of splenic small B-cell lymphomas. A study of 42 cases with a definitive diagnosis by the World Health Organization classification. Am. J. Clin. Pathol. 2003, 120, 335–347. [Google Scholar] [CrossRef]
- Bonfiglio, F.; Bruscaggin, A.; Guidetti, F.; di Bergamo, L.T.; Faderl, M.; Spina, V.; Condoluci, A.; Bonomini, L.; Forestieri, G.; Koch, R.; et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood 2022, 139, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Pettirossi, V.; Schiavoni, G.; Falini, B. Genomics of Hairy Cell Leukemia. J. Clin. Oncol. 2017, 35, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Dixon, J.G.; Le-Rademacher, J.; Hoster, E.; Hochster, H.S.; Hiddemann, W.; Marcus, R.; Kimby, E.; Herold, M.; Sebban, C.; et al. Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 2022, 139, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Al-Tourah, A.J.; Gill, K.K.; Chhanabhai, M.; Hoskins, P.J.; Klasa, R.J.; Savage, K.J.; Sehn, L.H.; Shenkier, T.N.; Gascoyne, R.D.; Connors, J.M. Population-based analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 5165–5169. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Primers 2019, 5, 83. [Google Scholar] [CrossRef]
- Green, M.R. Chromatin modifying gene mutations in follicular lymphoma. Blood 2018, 131, 595–604. [Google Scholar] [CrossRef]
- Küppers, R.; Stevenson, F.K. Critical influences on the pathogenesis of follicular lymphoma. Blood 2018, 131, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- von Keudell, G.; Salles, G. The role of tazemetostat in relapsed/refractory follicular lymphoma. Ther. Adv. Hematol. 2021, 12, 20406207211015882. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef] [Green Version]
- Mann, R.B.; Berard, C.W. Criteria for the cytologic subclassification of follicular lymphomas: A proposed alternative method. Hematol. Oncol. 1983, 1, 187–192. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. (Eds.) World Health Organization Classification of Haematopoietic Tumours. In Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001. [Google Scholar]
- Kansal, R. The World Health Organization (WHO) Classification of Tumors with Emphasis on the Classification of Hematolymphoid Neoplasms. In Precision Medicine: Where Are We and Where Are We Going? Nova Science Publishers, Inc.: New York, NY, USA, 2023; pp. 315–416. [Google Scholar]
- Feller, A.C.; Diebold, J. History of lymphoma classification. In Histopathology of Nodal and Extranodal Non-Hodgkin’s Lymphomas Based on the WHO Classification, 3rd ed.; Completely Revised and Updated Edition; Keller, A.C., Diebold, J., Paulli, M., Le Tourneau, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 1–13. [Google Scholar]
- Metter, G.E.; Nathwani, B.N.; Burke, J.S.; Winberg, C.D.; Mann, R.B.; Barcos, M.; Kjeldsberg, C.R.; Whitcomb, C.C.; Dixon, D.O.; Miller, T.P.; et al. Morphological subclassification of follicular lymphoma: Variability of diagnoses among hematopathologists, a collaborative study between the Repository Center and Pathology Panel for Lymphoma Clinical Studies. J. Clin. Oncol. 1985, 3, 25–38. [Google Scholar] [CrossRef]
- Cree, I.A.; Tan, P.H.; Travis, W.D.; Wesseling, P.; Yagi, Y.; White, V.A.; Lokuhetty, D.; Scolyer, R.A. Counting mitoses: SI(ze) matters! Mod. Pathol. 2021, 34, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.M.; Li, H.; Braziel, R.M.; Spier, C.M.; Persky, D.O.; Dunlap, J.; LeBlanc, M.; Bartlett, N.; Leonard, J.P.; Smith, S.M.; et al. Impact of histological grading on survival in the SWOG S0016 follicular lymphoma cohort. Haematologica 2018, 103, e151–e153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, A.; Gooley, T.; Loeb, K.; Soma, L.; Smith, S.D.; Gopal, A.; Naresh, K.N. The impact of histological grade on outcomes in follicular lymphoma: An analysis of patients in the SEER database in the context of evolving disease classification and treatment. Br. J. Haematol. 2022, 199, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A., Jr.; Schafernak, K.T.; Geyer, J.T.; Kovach, A.E.; Ghandi, M.; Gratzinger, D.; Roth, C.G.; Paxton, C.N.; Kim, S.; Namgyal, C.; et al. Pediatric-type nodal follicular lymphoma: A biologically distinct lymphoma with frequent MAPK pathway mutations. Blood 2016, 128, 1093–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Bosman, F.T., Lakhani, S.R., Jaffe, E.S., Ohgaki, H., Eds.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Schmidt, J.; Gong, S.; Marafioti, T.; Mankel, B.; Gonzalez-Farre, B.; Balagué, O.; Mozos, A.; Cabeçadas, J.; van der Walt, J.; Hoehn, D.; et al. Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of TNFRSF14 gene. Blood 2016, 128, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Salmeron-Villalobos, J.; Egan, C.; Borgmann, V.; Müller, I.; Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Nann, D.; Balagué, O.; López-Guerra, M.; Colomer, D.; et al. A unifying hypothesis for PNMZL and PTFL: Morphological variants with a common molecular profile. Blood Adv. 2022, 6, 4661–4674. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Abla, O.; Arias Padilla, L.; Beishuizen, A.; Burke, G.A.A.; Brugières, L.; Bruneau, J.; Burkhardt, B.; d’Amore, E.S.G.; Klapper, W.; et al. Rare non-Hodgkin lymphoma of childhood and adolescence: A consensus diagnostic and therapeutic approach to pediatric-type follicular lymphoma, marginal zone lymphoma, and nonanaplastic peripheral T-cell lymphoma. Pediatr. Blood Cancer 2020, 67, e28416. [Google Scholar] [CrossRef]
- Nann, D.; Ramis-Zaldivar, J.E.; Müller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef]
- Lones, M.A.; Raphael, M.; McCarthy, K.; Wotherspoon, A.; Terrier-Lacombe, M.J.; Ramsay, A.D.; Maclennan, K.; Cairo, M.S.; Gerrard, M.; Michon, J.; et al. Primary follicular lymphoma of the testis in children and adolescents. J. Pediatr. Hematol. Oncol. 2012, 34, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Kansal, R. In Situ Mantle Cell Neoplasia. Compendium of Cancer Genome Aberrations (CCGA), Cancer Genomics Consortium (CGC). Available online: https://www.ccga.io/index.php/In_Situ_Mantle_Cell_Neoplasia (accessed on 26 July 2022).
- Bermudez, G.; González de Villambrosía, S.; Martínez-López, A.; Batlle, A.; Revert-Arce, J.B.; Cereceda Company, L.; Ortega Bezanilla, C.; Piris, M.A.; Montes-Moreno, S. Incidental and Isolated Follicular Lymphoma In Situ and Mantle Cell Lymphoma In Situ Lack Clinical Significance. Am. J. Surg. Pathol. 2016, 40, 943–949. [Google Scholar] [CrossRef]
- Adam, P.; Schiefer, A.I.; Prill, S.; Henopp, T.; Quintanilla-Martínez, L.; Bösmüller, H.C.; Chott, A.; Fend, F. Incidence of preclinical manifestations of mantle cell lymphoma and mantle cell lymphoma in situ in reactive lymphoid tissues. Mod. Pathol. 2012, 25, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal-Cuenca, A.; Sua, L.F.; Silva, N.M.; Pittaluga, S.; Royo, C.; Song, J.Y.; Sargent, R.L.; Espinet, B.; Climent, F.; Jacobs, S.A.; et al. In situ mantle cell lymphoma: Clinical implications of an incidental finding with indolent clinical behavior. Haematologica 2012, 97, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Healey, B.M.; Pinkus, G.S.; Kuo, F.C.; Dal Cin, P.; Kutok, J.L. Mantle cell lymphoma arising within primary nodal marginal zone lymphoma: A unique presentation of two uncommon B-cell lymphoproliferative disorders. Cancer Genet. Cytogenet. 2006, 171, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Aqel, N.; Barker, F.; Patel, K.; Naresh, K.N. In-situ mantle cell lymphoma--a report of two cases. Histopathology 2008, 52, 256–260. [Google Scholar] [CrossRef]
- Roullet, M.R.; Martinez, D.; Ma, L.; Fowler, M.H.; McPhail, E.D.; Judkins, A.; Arber, D.A.; Bagg, A. Coexisting follicular and mantle cell lymphoma with each having an in situ component: A novel, curious, and complex consultation case of coincidental, composite, colonizing lymphoma. Am. J. Clin. Pathol. 2010, 133, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Demurtas, A.; Aliberti, S.; Bonello, L.; Di Celle, P.F.; Cavaliere, C.; Barreca, A.; Novero, D.; Stacchini, A. Usefulness of multiparametric flow cytometry in detecting composite lymphoma: Study of 17 cases in a 12-year period. Am. J. Clin. Pathol. 2011, 135, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Papathomas, T.G.; Venizelos, I.; Dunphy, C.H.; Said, J.W.; Wang, M.L.; Campo, E.; Swerdlow, S.H.; Chan, J.C.; Bueso-Ramos, C.E.; Weisenburger, D.D.; et al. Mantle cell lymphoma as a component of composite lymphoma: Clinicopathologic parameters and biologic implications. Hum. Pathol. 2012, 43, 467–480. [Google Scholar] [CrossRef]
- Subtil, A.; Xu, Z. Follicular lymphoma with composite in situ mantle cell neoplasia. Blood 2019, 133, 2460. [Google Scholar] [CrossRef]
- Bassarova, A.; Tierens, A.; Lauritzsen, G.F.; Fosså, A.; Delabie, J. Mantle cell lymphoma with partial involvement of the mantle zone: An early infiltration pattern of mantle cell lymphoma? Virchows Arch. 2008, 453, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Neto, A.G.; Oroszi, G.; Protiva, P.; Rose, M.; Shafi, N.; Torres, R. Colonic in situ mantle cell lymphoma. Ann. Diagn. Pathol. 2012, 16, 508–514. [Google Scholar] [CrossRef]
- Teixeira Mendes, L.S.; Wotherspoon, A. The relationship between overt and in-situ lymphoma: A retrospective study of follicular and mantle cell lymphoma cases. Histopathology 2016, 68, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Edlefsen, K.L.; Greisman, H.A.; Yi, H.S.; Mantei, K.M.; Fromm, J.R. Early lymph node involvement by mantle cell lymphoma limited to the germinal center: Report of a case with a novel “follicular in situ” growth pattern. Am. J. Clin. Pathol. 2011, 136, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Espinet, B.; Solé, F.; Pedro, C.; Garcia, M.; Bellosillo, B.; Salido, M.; Florensa, L.; Camacho, F.I.; Baró, T.; Lloreta, J.; et al. Clonal proliferation of cyclin D1-positive mantle lymphocytes in an asymptomatic patient: An early-stage event in the development or an indolent form of a mantle cell lymphoma? Hum. Pathol. 2005, 36, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Koletsa, T.; Markou, K.; Ouzounidou, S.; Tsiompanou, F.; Karkavelas, G.; Kostopoulos, I. In situ mantle cell lymphoma in the nasopharynx. Head Neck 2013, 35, E333–E337. [Google Scholar] [CrossRef]
- Dobrea, C.; Mihai, M.; Dănăilă, E.; Găman, A.; Coriu, D.; Ursuleac, I. “In situ” mantle cell lymphoma associated with hyaline-vascular Castleman disease. Rom. J. Morphol. Embryol. 2011, 52 (Suppl. S3), 1147–1151. [Google Scholar]
- Zanelli, M.; Stingeni, L.; Zizzo, M.; Martino, G.; Sanguedolce, F.; Marra, A.; Crescenzi, B.; Pileri, S.A.; Ascani, S. HHV8-Positive Castleman Disease and In Situ Mantle Cell Neoplasia within Dermatopathic Lymphadenitis, in Longstanding Psoriasis. Diagnostics 2021, 11, 1150. [Google Scholar] [CrossRef]
- Hirt, C.; Schüler, F.; Dölken, L.; Schmidt, C.A.; Dölken, G. Low prevalence of circulating t(11;14)(q13;q32)-positive cells in the peripheral blood of healthy individuals as detected by real-time quantitative PCR. Blood 2004, 104, 904–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambiar, M.; Raghavan, S.C. Prevalence and analysis of t(14;18) and t(11;14) chromosomal translocations in healthy Indian population. Ann. Hematol. 2010, 89, 35–43. [Google Scholar] [CrossRef]
- Lecluse, Y.; Lebailly, P.; Roulland, S.; Gac, A.C.; Nadel, B.; Gauduchon, P. t(11;14)-positive clones can persist over a long period of time in the peripheral blood of healthy individuals. Leukemia 2009, 23, 1190–1193. [Google Scholar] [CrossRef] [Green Version]
- Weisenburger, D.D.; Kim, H.; Rappaport, H. Mantle-zone lymphoma: A follicular variant of intermediate lymphocytic lymphoma. Cancer 1982, 49, 1429–1438. [Google Scholar] [CrossRef]
- Weisenburger, D.D.; Nathwani, B.N.; Diamond, L.W.; Winberg, C.D.; Rappaport, H. Malignant lymphoma, intermediate lymphocytic type: A clinicopathologic study of 42 cases. Cancer 1981, 48, 1415–1425. [Google Scholar] [CrossRef]
- Williams, M.E.; Westermann, C.D.; Swerdlow, S.H. Genotypic characterization of centrocytic lymphoma: Frequent rearrangement of the chromosome 11 bcl-1 locus. Blood 1990, 76, 1387–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, L.J.; Van Krieken, J.H.; Jaffe, E.S.; Raffeld, M. Association of bcl-1 rearrangements with lymphocytic lymphoma of intermediate differentiation. Blood 1990, 76, 2086–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, P.M.; Chan, J.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Gatter, K.; Grogan, T.M.; Harris, N.L.; Isaacson, P.G.; Jaffe, E.S.; et al. Mantle cell lymphoma. A proposal for unification of morphologic, immunologic, and molecular data. Am. J. Surg. Pathol. 1992, 16, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O.; Longo, D.L. Mantle-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 2495–2506. [Google Scholar] [CrossRef]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic mutations and features of mantle cell lymphoma: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [Google Scholar] [CrossRef]
- Jain, P.; Wang, M.L. Mantle cell lymphoma in 2022-A comprehensive update on molecular pathogenesis, risk stratification, clinical approach, and current and novel treatments. Am. J. Hematol. 2022, 97, 638–656. [Google Scholar] [CrossRef]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Analysis of 153 115 patients with hematological malignancies refines the spectrum of familial risk. Blood 2019, 134, 960–969. [Google Scholar] [CrossRef]
- Tort, F.; Camacho, E.; Bosch, F.; Harris, N.L.; Montserrat, E.; Campo, E. Familial lymphoid neoplasms in patients with mantle cell lymphoma. Haematologica 2004, 89, 314–319. [Google Scholar]
- Hangaishi, A.; Ogawa, S.; Qiao, Y.; Wang, L.; Hosoya, N.; Yuji, K.; Imai, Y.; Takeuchi, K.; Miyawaki, S.; Hirai, H. Mutations of Chk2 in primary hematopoietic neoplasms. Blood 2002, 99, 3075–3077. [Google Scholar] [CrossRef] [Green Version]
- Camacho, E.; Hernández, L.; Hernández, S.; Tort, F.; Bellosillo, B.; Beà, S.; Bosch, F.; Montserrat, E.; Cardesa, A.; Fernández, P.L.; et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood 2002, 99, 238–244. [Google Scholar] [CrossRef]
- Nadeu, F.; Martin-Garcia, D.; Clot, G.; Díaz-Navarro, A.; Duran-Ferrer, M.; Navarro, A.; Vilarrasa-Blasi, R.; Kulis, M.; Royo, R.; Gutiérrez-Abril, J.; et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood 2020, 136, 1419–1432. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Chen, W.; Ding, N.; Liu, W.; Xie, Y.; Wang, Y.; Zhu, J.; Zeng, C. Germline variants of DNA repair genes in early onset mantle cell lymphoma. Oncogene 2021, 40, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Treon, S.P.; Hunter, Z.R. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood 2013, 121, 4434–4436. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Buhrlage, S.J.; Tan, L.; Liu, X.; Chen, J.; Xu, L.; Tsakmaklis, N.; Chen, J.G.; Patterson, C.J.; Brown, J.R.; et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 2016, 127, 3237–3252. [Google Scholar] [CrossRef] [Green Version]
- Hunter, Z.R.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Manning, R.J.; Tripsas, C.; Patterson, C.J.; Sheehy, P.; et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014, 123, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Treon, S.P.; Cao, Y.; Xu, L.; Yang, G.; Liu, X.; Hunter, Z.R. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014, 123, 2791–2796. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.J.; Garcia-Sanz, R.; Hatjiharissi, E.; Kyle, R.A.; Leleu, X.; McMaster, M.; Merlini, G.; Minnema, M.C.; Morra, E.; Owen, R.G.; et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström Macroglobulinaemia: A Task Force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br. J. Haematol. 2016, 175, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treon, S.P.; Gustine, J.; Xu, L.; Manning, R.J.; Tsakmaklis, N.; Demos, M.; Meid, K.; Guerrera, M.L.; Munshi, M.; Chan, G.; et al. MYD88 wild-type Waldenstrom Macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br. J. Haematol. 2018, 180, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Jimenez, C.; Chan, G.G.; Chen, J.; Liu, X.; Munshi, M.; et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018, 2, 2937–2946. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Treon, S.P. Epigenomics in Waldenström macroglobulinemia. Blood 2020, 136, 527–529. [Google Scholar] [CrossRef]
- Kofides, A.; Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Munshi, M.; Liu, X.; Guerrera, M.L.; Leventoff, C.R.; White, T.P.; et al. Diagnostic Next-generation Sequencing Frequently Fails to Detect MYD88L265P in Waldenström Macroglobulinemia. Hemasphere 2021, 5, e624. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Jia, M.N.; Cai, H.; Qiu, Y.; Zhou, D.B.; Li, J.; Cao, X.X. Detection of the MYD88L265P and CXCR4S338X mutations by cell-free DNA in Waldenström macroglobulinemia. Ann. Hematol. 2020, 99, 1763–1769. [Google Scholar] [CrossRef]
- Demos, M.G.; Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Kofides, A.; Munshi, M.; Liu, X.; Guerrera, M.L.; Leventoff, C.R.; White, T.P.; et al. Cell-free DNA analysis for detection of MYD88L265P and CXCR4S338X mutations in Waldenström macroglobulinemia. Am. J. Hematol. 2021, 96, E250–E253. [Google Scholar] [CrossRef]
- Varettoni, M.; Boveri, E.; Zibellini, S.; Tedeschi, A.; Candido, C.; Ferretti, V.V.; Rizzo, E.; Doni, E.; Merli, M.; Farina, L.; et al. Clinical and molecular characteristics of lymphoplasmacytic lymphoma not associated with an IgM monoclonal protein: A multicentric study of the Rete Ematologica Lombarda (REL) network. Am. J. Hematol. 2019, 94, 1193–1199. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.; Hanzis, C.; Ioakimidis, L.; Patterson, C.J.; Manning, R.J.; Sheehy, P.; Turnbull, B.; Hunter, Z.R. Familial disease predisposition impacts treatment outcome in patients with Waldenström macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 433–437. [Google Scholar] [CrossRef]
- Treon, S.P.; Hunter, Z.R.; Aggarwal, A.; Ewen, E.P.; Masota, S.; Lee, C.; Santos, D.D.; Hatjiharissi, E.; Xu, L.; Leleu, X.; et al. Characterization of familial Waldenstrom’s macroglobulinemia. Ann. Oncol. 2006, 17, 488–494. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Shi, J.; Chiarini, M.; Perilla-Glen, A.; Manier, S.; Glavey, S.; Aljawai, Y.; Mishima, Y.; Kawano, Y.; et al. Exome sequencing reveals recurrent germ line variants in patients with familial Waldenström macroglobulinemia. Blood 2016, 127, 2598–2606. [Google Scholar] [CrossRef] [Green Version]
- Abboudi, Z.; Patel, K.; Naresh, K.N. Cyclin D1 expression in typical chronic lymphocytic leukaemia. Eur. J. Haematol. 2009, 83, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Miquel, R.; Krenacs, L.; Sorbara, L.; Raffeld, M.; Jaffe, E.S. Primary nodal marginal zone lymphomas of splenic and MALT type. Am. J. Surg. Pathol. 1999, 23, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Weiss, L.M.; Chang, K.L.; Arber, D.A. Frequency of CD43 expression in non-Hodgkin lymphoma. A survey of 742 cases and further characterization of rare CD43+ follicular lymphomas. Am. J. Clin. Pathol. 1999, 111, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrath, I. Epidemiology: Clues to the pathogenesis of Burkitt lymphoma. Br. J. Haematol. 2012, 156, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Liu, H.; Miranda, R.N.; Golardi, N.; Billah, S.; Medeiros, L.J.; Jaso, J.M. Burkitt lymphoma presenting as a mass in the thyroid gland: A clinicopathologic study of 7 cases and review of the literature. Hum. Pathol. 2016, 56, 101–108. [Google Scholar] [CrossRef]
- Lenze, D.; Leoncini, L.; Hummel, M.; Volinia, S.; Liu, C.G.; Amato, T.; De Falco, G.; Githanga, J.; Horn, H.; Nyagol, J.; et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia 2011, 25, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Gopal, S.; Gross, T.G. How I treat Burkitt lymphoma in children, adolescents, and young adults in sub-Saharan Africa. Blood 2018, 132, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Moormann, A.M.; Bailey, J.A. Malaria—How this parasitic infection aids and abets EBV-associated Burkitt lymphomagenesis. Curr. Opin. Virol. 2016, 20, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Pelicci, P.G.; Knowles, D.M., 2nd; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.J.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med. 2006, 354, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellan, C.; Lazzi, S.; Hummel, M.; Palummo, N.; de Santi, M.; Amato, T.; Nyagol, J.; Sabattini, E.; Lazure, T.; Pileri, S.A.; et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood 2005, 106, 1031–1036. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Richter, J.; John, K.; Staiger, A.M.; Rosenwald, A.; Kurz, K.; Michgehl, U.; Ott, G.; Franzenburg, S.; Kohler, C.; Finger, J.; et al. Epstein-Barr virus status of sporadic Burkitt lymphoma is associated with patient age and mutational features. Br. J. Haematol. 2022, 196, 681–689. [Google Scholar] [CrossRef]
- Thomas, N.; Dreval, K.; Gerhard, D.S.; Hilton, L.K.; Abramson, J.S.; Ambinder, R.F.; Barta, S.; Bartlett, N.L.; Bethony, J.; Bhatia, K.; et al. Genetic subgroups inform on pathobiology in adult and pediatric Burkitt lymphoma. Blood 2023, 141, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Naresh, K.N.; Lazzi, S.; Santi, R.; Granai, M.; Onyango, N.; Leoncini, L. A refined approach to the diagnosis of Burkitt lymphoma in a resource-poor setting. Histopathology 2022, 80, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Deeb, G.; Barcos, M.; Wetzler, M.; Brecher, M.L.; Block, A.W.; Stewart, C.C. Precursor B lymphoblastic leukemia with surface light chain immunoglobulin restriction: A report of 15 patients. Am. J. Clin. Pathol. 2004, 121, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W. Relapsed/refractory diffuse large B-cell lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Davies, A. Tailoring front-line therapy in diffuse large B-cell lymphoma: Who should we treat differently? Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [Green Version]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Jiang, Y.; Dominguez, P.M.; Melnick, A.M. The many layers of epigenetic dysfunction in B-cell lymphomas. Curr. Opin. Hematol. 2016, 23, 377–384. [Google Scholar] [CrossRef]
- Lunning, M.A.; Green, M.R. Mutation of chromatin modifiers; An emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J. 2015, 5, e361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Young, R.M.; Staudt, L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Morin, R.D.; Arthur, S.E.; Hodson, D.J. Molecular profiling in diffuse large B-cell lymphoma: Why so many types of subtypes? Br. J. Haematol. 2022, 196, 814–829. [Google Scholar] [CrossRef]
- Cucco, F.; Barrans, S.; Sha, C.; Clipson, A.; Crouch, S.; Dobson, R.; Chen, Z.; Thompson, J.S.; Care, M.A.; Cummin, T.; et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia 2020, 34, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.W.; King, R.L.; Staiger, A.M.; Ben-Neriah, S.; Jiang, A.; Horn, H.; Mottok, A.; Farinha, P.; Slack, G.W.; Ennishi, D.; et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 2018, 131, 2060–2064. [Google Scholar] [CrossRef]
- Rosenwald, A.; Bens, S.; Advani, R.; Barrans, S.; Copie-Bergman, C.; Elsensohn, M.H.; Natkunam, Y.; Calaminici, M.; Sander, B.; Baia, M.; et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2019, 37, 3359–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, C.; Barrans, S.; Cucco, F.; Bentley, M.A.; Care, M.A.; Cummin, T.; Kennedy, H.; Thompson, J.S.; Uddin, R.; Worrillow, L.; et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J. Clin. Oncol. 2019, 37, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Ennishi, D.; Jiang, A.; Boyle, M.; Collinge, B.; Grande, B.M.; Ben-Neriah, S.; Rushton, C.; Tang, J.; Thomas, N.; Slack, G.W.; et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 190–201. [Google Scholar] [CrossRef]
- Salaverria, I.; Martin-Guerrero, I.; Wagener, R.; Kreuz, M.; Kohler, C.W.; Richter, J.; Pienkowska-Grela, B.; Adam, P.; Burkhardt, B.; Claviez, A.; et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 2014, 123, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Wagener, R.; Seufert, J.; Raimondi, F.; Bens, S.; Kleinheinz, K.; Nagel, I.; Altmüller, J.; Thiele, H.; Hübschmann, D.; Kohler, C.W.; et al. The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood 2019, 133, 962–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Salmeron-Villalobos, J.; Balagué, O.; Celis, V.; Verdu-Amoros, J.; Nadeu, F.; Sábado, C.; Ferrández, A.; Garrido, M.; et al. Burkitt-like lymphoma with 11q aberration: A germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica 2019, 104, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Kalmbach, S.; Wagener, R.; Staiger, A.M.; Hüttl, K.; Mottok, A.; Bens, S.; Traverse-Glehen, A.; Fontaine, J.; Siebert, R.; et al. A Diagnostic Approach to the Identification of Burkitt-like Lymphoma With 11q Aberration in Aggressive B-Cell Lymphomas. Am. J. Surg. Pathol. 2021, 45, 356–364. [Google Scholar] [CrossRef]
- Yu, Y.T.; Takeuchi, K.; Baba, S.; Chang, K.C. Morphologically Suspected Burkitt-like Lymphoma With 11q Aberrations Confirmed by Fluorescence In Situ Hybridization. Am. J. Surg. Pathol. 2022, 46, 576–577. [Google Scholar] [CrossRef]
- Rymkiewicz, G.; Grygalewicz, B.; Chechlinska, M.; Blachnio, K.; Bystydzienski, Z.; Romejko-Jarosinska, J.; Woroniecka, R.; Zajdel, M.; Domanska-Czyz, K.; Martin-Garcia, D.; et al. A comprehensive flow-cytometry-based immunophenotypic characterization of Burkitt-like lymphoma with 11q aberration. Mod. Pathol. 2018, 31, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Arnold, L.M.; D’Agostino, E.; Thomas, A.A.; DeWitt, J.C. Tumors of the Central Nervous System. In Precision Medicine: Where Are We and Where Are We Going? Kansal, R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2023. [Google Scholar]
- Shiels, M.S.; Pfeiffer, R.M.; Besson, C.; Clarke, C.A.; Morton, L.M.; Nogueira, L.; Pawlish, K.; Yanik, E.L.; Suneja, G.; Engels, E.A. Trends in primary central nervous system lymphoma incidence and survival in the U.S. Br. J. Haematol. 2016, 174, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Ferreri, A.J.M. Secondary CNS lymphoma: The poisoned needle in the haystack. Ann. Oncol. 2017, 28, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.; Kim, S.J.; Kim, K.; Lee, J.I.; Lim, D.H.; Lee, D.J.; Baek, K.K.; Lee, H.Y.; Han, B.; Uhm, J.E.; et al. Primary CNS lymphoma other than DLBCL: A descriptive analysis of clinical features and treatment outcomes. Ann. Hematol. 2011, 90, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Brunn, A.; Bentink, S.; Basso, K.; Lim, W.K.; Klapper, W.; Schaller, C.; Reifenberger, G.; Rubenstein, J.; Wiestler, O.D.; et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia 2008, 22, 400–405. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Purschke, F.; Küppers, R.; Deckert, M. Immunoglobulin repertoire of primary lymphomas of the central nervous system. J. Neuropathol. Exp. Neurol. 2014, 73, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Küppers, R.; Schlüter, D.; Spieker, T.; Van Roost, D.; Schaller, C.; Reifenberger, G.; Wiestler, O.D.; Deckert-Schlüter, M. Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4-34 gene segment. Am. J. Pathol. 1999, 155, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Belhouachi, N.; Xochelli, A.; Boudjoghra, M.; Lesty, C.; Cassoux, N.; Fardeau, C.; Tran, T.H.C.; Choquet, S.; Sarker, B.; Houillier, C.; et al. Primary vitreoretinal lymphomas display a remarkably restricted immunoglobulin gene repertoire. Blood Adv. 2020, 4, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Radke, J.; Ishaque, N.; Koll, R.; Gu, Z.; Schumann, E.; Sieverling, L.; Uhrig, S.; Hübschmann, D.; Toprak, U.H.; López, C.; et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat. Commun. 2022, 13, 2558. [Google Scholar] [CrossRef]
- Casellas, R.; Basu, U.; Yewdell, W.T.; Chaudhuri, J.; Robbiani, D.F.; Di Noia, J.M. Mutations, kataegis and translocations in B cells: Understanding AID promiscuous activity. Nat. Rev. Immunol. 2016, 16, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Deckert, M.; Brunn, A.; Montesinos-Rongen, M.; Terreni, M.R.; Ponzoni, M. Primary lymphoma of the central nervous system—A diagnostic challenge. Hematol. Oncol. 2014, 32, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E.; Damato, B. Understanding intraocular lymphomas. Clin. Exp. Ophthalmol. 2008, 36, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.S.; Johnston, P.B.; Nowakowski, G.S.; Castellino, A.; Raja, H. The diagnosis and treatment of primary vitreoretinal lymphoma: A review. Int. J. Retin. Vitr. 2018, 4, 18. [Google Scholar] [CrossRef]
- Zuckerman, D.; Seliem, R.; Hochberg, E. Intravascular lymphoma: The oncologist’s “great imitator”. Oncologist 2006, 11, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonkem, E.; Dayawansa, S.; Stroberg, E.; Lok, E.; Bricker, P.C.; Kirmani, B.; Wong, E.T.; Huang, J.H. Neurological presentations of intravascular lymphoma (IVL): Meta-analysis of 654 patients. BMC Neurol. 2016, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Kajimoto, K.; Taniwaki, M.; Miura, I.; Hatta, Y.; Hashizume, Y.; Watanabe, M.; Shiraishi, T.; Nakamura, S. Intravascular large B-cell lymphoma presenting with mass lesions in the central nervous system: A report of five cases. Pathol. Int. 2004, 54, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Campo, E.; Nakamura, S. Intravascular large B-cell lymphoma: A chameleon with multiple faces and many masks. Blood 2018, 132, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kiyoi, H. Current progress and future perspectives of research on intravascular large B-cell lymphoma. Cancer Sci. 2021, 112, 3953–3961. [Google Scholar] [CrossRef]
- Fonkem, E.; Lok, E.; Robison, D.; Gautam, S.; Wong, E.T. The natural history of intravascular lymphomatosis. Cancer Med. 2014, 3, 1010–1024. [Google Scholar] [CrossRef]
- Geer, M.; Roberts, E.; Shango, M.; Till, B.G.; Smith, S.D.; Abbas, H.; Hill, B.T.; Kaplan, J.; Barr, P.M.; Caimi, P.; et al. Multicentre retrospective study of intravascular large B-cell lymphoma treated at academic institutions within the United States. Br. J. Haematol. 2019, 186, 255–262. [Google Scholar] [CrossRef]
- Seegobin, K.; Li, Z.; Alhaj Moustafa, M.; Majeed, U.; Wang, J.; Jiang, L.; Kuhlman, J.; Menke, D.; Li, K.; Kharfan-Dabaja, M.A.; et al. Clinical characteristics, prognostic indicators, and survival outcomes in intravascular lymphoma: Mayo Clinic experience (2003–2018). Am. J. Hematol. 2022, 97, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Yoshida, K.; Suzuki, Y.; Iriyama, C.; Inoue, Y.; Sanada, M.; Kataoka, K.; Yuge, M.; Takagi, Y.; Kusumoto, S.; et al. Frequent genetic alterations in immune checkpoint-related genes in intravascular large B-cell lymphoma. Blood 2021, 137, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Suehara, Y.; Sakata-Yanagimoto, M.; Hattori, K.; Nanmoku, T.; Itoh, T.; Kaji, D.; Yamamoto, G.; Abe, Y.; Narita, K.; Takeuchi, M.; et al. Liquid biopsy for the identification of intravascular large B-cell lymphoma. Haematologica 2018, 103, e241–e244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.H.; Cleton-Jansen, A.M.; Somers, S.F.; Veelken, H.; van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Verdun, R.E.; Lossos, I.S. Low LIM-domain only 2 (LMO2) expression in aggressive B cell lymphoma correlates with MYC and MYC/BCL2 rearrangements, especially in germinal center cell-type tumors. Leuk. Lymphoma 2021, 62, 2547–2550. [Google Scholar] [CrossRef]
- Salaverria, I.; Philipp, C.; Oschlies, I.; Kohler, C.W.; Kreuz, M.; Szczepanowski, M.; Burkhardt, B.; Trautmann, H.; Gesk, S.; Andrusiewicz, M.; et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011, 118, 139–147. [Google Scholar] [CrossRef]
- Chen, L.; Al-Kzayer, L.F.; Liu, T.; Kobayashi, N.; Nakazawa, Y.; Koike, K. IFR4/MUM1-positive lymphoma in Waldeyer ring with co-expression of CD5 and CD10. Pediatr. Blood Cancer 2017, 64, 311–314. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Seto, M.; Okamoto, M.; Ichinohasama, R.; Nakamura, N.; Yoshino, T.; Suzumiya, J.; Murase, T.; Miura, I.; Akasaka, T.; et al. De novo CD5+ diffuse large B-cell lymphoma: A clinicopathologic study of 109 patients. Blood 2002, 99, 815–821. [Google Scholar] [CrossRef]
- Parvin, S.; Ramirez-Labrada, A.; Aumann, S.; Lu, X.; Weich, N.; Santiago, G.; Cortizas, E.M.; Sharabi, E.; Zhang, Y.; Sanchez-Garcia, I.; et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell 2019, 36, 237–249.e6. [Google Scholar] [CrossRef]
- Natkunam, Y.; Zhao, S.; Mason, D.Y.; Chen, J.; Taidi, B.; Jones, M.; Hammer, A.S.; Hamilton Dutoit, S.; Lossos, I.S.; Levy, R. The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 2007, 109, 1636–1642. [Google Scholar] [CrossRef] [Green Version]
- Piña-Oviedo, S.; Moran, C.A. Primary Mediastinal Nodal and Extranodal Non-Hodgkin Lymphomas: Current Concepts, Historical Evolution, and Useful Diagnostic Approach: Part 1. Adv. Anat. Pathol. 2019, 26, 346–370. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, T.; Jaffe, E.S.; Delsol, G.; deWolf-Peeters, C.; Gascoyne, R.D.; Georgii, A.; Harris, N.L.; Kadin, M.E.; MacLennan, K.A.; Poppema, S.; et al. Workshop report on Hodgkin’s disease and related diseases (‘grey zone’ lymphoma). Ann. Oncol. 1998, 9 (Suppl. S5), S31–S38. [Google Scholar] [CrossRef] [PubMed]
- Traverse-Glehen, A.; Pittaluga, S.; Gaulard, P.; Sorbara, L.; Alonso, M.A.; Raffeld, M.; Jaffe, E.S. Mediastinal gray zone lymphoma: The missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am. J. Surg. Pathol. 2005, 29, 1411–1421. [Google Scholar] [CrossRef]
- García, J.F.; Mollejo, M.; Fraga, M.; Forteza, J.; Muniesa, J.A.; Pérez-Guillermo, M.; Pérez-Seoane, C.; Rivera, T.; Ortega, P.; Piris, M.A. Large B-cell lymphoma with Hodgkin’s features. Histopathology 2005, 47, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Dunleavy, K.; Eberle, F.C.; Pittaluga, S.; Wilson, W.H.; Jaffe, E.S. Primary mediastinal large B-cell lymphoma, classic Hodgkin lymphoma presenting in the mediastinum, and mediastinal gray zone lymphoma: What is the oncologist to do? Curr. Hematol. Malig. Rep. 2011, 6, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.K.; Levine, A.; Taylor, C.R.; Boswell, W.; Rossman, S.; Feinstein, D.I.; Lukes, R.J. Primary mediastinal lymphoma in adults. Am. J. Med. 1980, 68, 509–514. [Google Scholar] [CrossRef]
- Cazals-Hatem, D.; Lepage, E.; Brice, P.; Ferrant, A.; d’Agay, M.F.; Baumelou, E.; Brière, J.; Blanc, M.; Gaulard, P.; Biron, P.; et al. Primary mediastinal large B-cell lymphoma. A clinicopathologic study of 141 cases compared with 916 nonmediastinal large B-cell lymphomas, a GELA (“Groupe d’Etude des Lymphomes de l’Adulte”) study. Am. J. Surg. Pathol. 1996, 20, 877–888. [Google Scholar] [CrossRef]
- Dunleavy, K. Primary mediastinal B-cell lymphoma: Biology and evolving therapeutic strategies. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Isaacson, P.G.; Norton, A.J.; Addis, B.J. The human thymus contains a novel population of B lymphocytes. Lancet 1987, 2, 1488–1491. [Google Scholar] [CrossRef]
- Hofmann, W.J.; Momburg, F.; Möller, P. Thymic medullary cells expressing B lymphocyte antigens. Hum. Pathol. 1988, 19, 1280–1287. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Monti, S.; Kutok, J.L.; Cattoretti, G.; Neuberg, D.; De Leval, L.; Kurtin, P.; Dal Cin, P.; Ladd, C.; Feuerhake, F.; et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef] [Green Version]
- Joos, S.; Otaño-Joos, M.I.; Ziegler, S.; Brüderlein, S.; du Manoir, S.; Bentz, M.; Möller, P.; Lichter, P. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 1996, 87, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Küpper, M.; Ohl, S.; von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Möller, P.; Pfreundschuh, M.; Trümper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Bentz, M.; Barth, T.F.; Brüderlein, S.; Bock, D.; Schwerer, M.J.; Baudis, M.; Joos, S.; Viardot, A.; Feller, A.C.; Müller-Hermelink, H.K.; et al. Gain of chromosome arm 9p is characteristic of primary mediastinal B-cell lymphoma (MBL): Comprehensive molecular cytogenetic analysis and presentation of a novel MBL cell line. Genes Chromosomes Cancer 2001, 30, 393–401. [Google Scholar] [CrossRef]
- Oschlies, I.; Burkhardt, B.; Salaverria, I.; Rosenwald, A.; d’Amore, E.S.; Szczepanowski, M.; Koch, K.; Hansmann, M.L.; Stein, H.; Möller, P.; et al. Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in children. Haematologica 2011, 96, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Rao, S.; Dhawan, S.; Bhalla, S.; Kumar, A.; Chopra, P. Primary mediastinal lymphomas, their morphological features and comparative evaluation. Lung India 2017, 34, 19–24. [Google Scholar] [CrossRef]
- al-Sharabati, M.; Chittal, S.; Duga-Neulat, I.; Laurent, G.; Mazerolles, C.; al-Saati, T.; Brousset, P.; Delsol, G. Primary anterior mediastinal B-cell lymphoma. A clinicopathologic and immunohistochemical study of 16 cases. Cancer 1991, 67, 2579–2587. [Google Scholar] [CrossRef]
- Yuan, J.; Wright, G.; Rosenwald, A.; Steidl, C.; Gascoyne, R.D.; Connors, J.M.; Mottok, A.; Weisenburger, D.D.; Greiner, T.C.; Fu, K.; et al. Identification of Primary Mediastinal Large B-cell Lymphoma at Nonmediastinal Sites by Gene Expression Profiling. Am. J. Surg. Pathol. 2015, 39, 1322–1330. [Google Scholar] [CrossRef]
- Wang, Y.; Wenzl, K.; Manske, M.K.; Asmann, Y.W.; Sarangi, V.; Greipp, P.T.; Krull, J.E.; Hartert, K.; He, R.; Feldman, A.L.; et al. Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J. 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Savage, K.J. Primary mediastinal large B-cell lymphoma. Blood 2022, 140, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Saarinen, S.; Kaasinen, E.; Karjalainen-Lindsberg, M.L.; Vesanen, K.; Aavikko, M.; Katainen, R.; Taskinen, M.; Kytölä, S.; Leppä, S.; Hietala, M.; et al. Primary mediastinal large B-cell lymphoma segregating in a family: Exome sequencing identifies MLL as a candidate predisposition gene. Blood 2013, 121, 3428–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilichowska, M.; Pittaluga, S.; Ferry, J.A.; Hemminger, J.; Chang, H.; Kanakry, J.A.; Sehn, L.H.; Feldman, T.; Abramson, J.S.; Kritharis, A.; et al. Clinicopathologic consensus study of gray zone lymphoma with features intermediate between DLBCL and classical HL. Blood Adv. 2017, 1, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkozy, C.; Copie-Bergman, C.; Damotte, D.; Ben-Neriah, S.; Burroni, B.; Cornillon, J.; Lemal, R.; Golfier, C.; Fabiani, B.; Chassagne-Clément, C.; et al. Gray-zone Lymphoma Between cHL and Large B-Cell Lymphoma: A Histopathologic Series From the LYSA. Am. J. Surg. Pathol. 2019, 43, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, S.; Nicolae, A.; Wright, G.W.; Melani, C.; Roschewski, M.; Steinberg, S.; Huang, D.; Staudt, L.M.; Jaffe, E.S.; Wilson, W.H. Gene Expression Profiling of Mediastinal Gray Zone Lymphoma and Its Relationship to Primary Mediastinal B-cell Lymphoma and Classical Hodgkin Lymphoma. Blood Cancer Discov. 2020, 1, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; Pittaluga, S.; Nicolae, A.; Camphausen, K.; Shovlin, M.; Steinberg, S.M.; Roschewski, M.; Staudt, L.M.; Jaffe, E.S.; Dunleavy, K. A prospective study of mediastinal gray-zone lymphoma. Blood 2014, 124, 1563–1569. [Google Scholar] [CrossRef] [Green Version]
- Aussedat, G.; Traverse-Glehen, A.; Stamatoullas, A.; Molina, T.; Safar, V.; Laurent, C.; Michot, J.M.; Hirsch, P.; Nicolas-Virelizier, E.; Lamure, S.; et al. Composite and sequential lymphoma between classical Hodgkin lymphoma and primary mediastinal lymphoma/diffuse large B-cell lymphoma, a clinico-pathological series of 25 cases. Br. J. Haematol. 2020, 189, 244–256. [Google Scholar] [CrossRef]
- Eberle, F.C.; Salaverria, I.; Steidl, C.; Summers, T.A., Jr.; Pittaluga, S.; Neriah, S.B.; Rodriguez-Canales, J.; Xi, L.; Ylaya, K.; Liewehr, D.; et al. Gray zone lymphoma: Chromosomal aberrations with immunophenotypic and clinical correlations. Mod. Pathol. 2011, 24, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Sarkozy, C.; Chong, L.; Takata, K.; Chavez, E.A.; Miyata-Takata, T.; Duns, G.; Telenius, A.; Boyle, M.; Slack, G.W.; Laurent, C.; et al. Gene expression profiling of gray zone lymphoma. Blood Adv. 2020, 4, 2523–2535. [Google Scholar] [CrossRef]
- Sarkozy, C.; Hung, S.S.; Chavez, E.A.; Duns, G.; Takata, K.; Chong, L.C.; Aoki, T.; Jiang, A.; Miyata-Takata, T.; Telenius, A.; et al. Mutational landscape of gray zone lymphoma. Blood 2021, 137, 1765–1776. [Google Scholar] [CrossRef]
- Kansal, R.; Sait, S.N.; Block, A.W.; Ward, P.M.; Kelly, F.L.; Cheney, R.T.; Czuczman, M.; Brecher, M.L.; Barcos, M. Extra copies of chromosome 2 are a recurring aberration in ALK-negative lymphomas with anaplastic morphology. Mod. Pathol. 2005, 18, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansal, R.; Singleton, T.P.; Ross, C.W.; Finn, W.G.; Padmore, R.F.; Schnitzer, B. Follicular hodgkin lymphoma: A histopathologic study. Am. J. Clin. Pathol. 2002, 117, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Vose, J.; Armitage, J.; Weisenburger, D.; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- de Leval, L.; Rickman, D.S.; Thielen, C.; de Reynies, A.; Huang, Y.L.; Delsol, G.; Lamant, L.; Leroy, K.; Brière, J.; Molina, T.; et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 2007, 109, 4952–4963. [Google Scholar] [CrossRef] [Green Version]
- Agostinelli, C.; Hartmann, S.; Klapper, W.; Korkolopoulou, P.; Righi, S.; Marafioti, T.; Piccaluga, P.P.; Patsouris, E.; Hansmann, M.L.; Lennert, K.; et al. Peripheral T cell lymphomas with follicular T helper phenotype: A new basket or a distinct entity? Revising Karl Lennert’s personal archive. Histopathology 2011, 59, 679–691. [Google Scholar] [CrossRef]
- Mourad, N.; Mounier, N.; Brière, J.; Raffoux, E.; Delmer, A.; Feller, A.; Meijer, C.J.; Emile, J.F.; Bouabdallah, R.; Bosly, A.; et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood 2008, 111, 4463–4470. [Google Scholar] [CrossRef]
- Chiba, S.; Sakata-Yanagimoto, M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia 2020, 34, 2592–2606. [Google Scholar] [CrossRef]
- Huang, Y.; Moreau, A.; Dupuis, J.; Streubel, B.; Petit, B.; Le Gouill, S.; Martin-Garcia, N.; Copie-Bergman, C.; Gaillard, F.; Qubaja, M.; et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 682–690. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef] [Green Version]
- Ghione, P.; Faruque, P.; Mehta-Shah, N.; Seshan, V.; Ozkaya, N.; Bhaskar, S.; Yeung, J.; Spinner, M.A.; Lunning, M.; Inghirami, G.; et al. T follicular helper phenotype predicts response to histone deacetylase inhibitors in relapsed/refractory peripheral T-cell lymphoma. Blood Adv. 2020, 4, 4640–4647. [Google Scholar] [CrossRef] [PubMed]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef]
- Krug, A.; Tari, G.; Saidane, A.; Gaulard, P.; Ricci, J.E.; Lemonnier, F.; Verhoeyen, E. Novel T Follicular Helper-like T-Cell Lymphoma Therapies: From Preclinical Evaluation to Clinical Reality. Cancers 2022, 14, 2392. [Google Scholar] [CrossRef]
- Attygalle, A.; Al-Jehani, R.; Diss, T.C.; Munson, P.; Liu, H.; Du, M.Q.; Isaacson, P.G.; Dogan, A. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood 2002, 99, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attygalle, A.D.; Cabeçadas, J.; Gaulard, P.; Jaffe, E.S.; de Jong, D.; Ko, Y.H.; Said, J.; Klapper, W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward—Report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology 2014, 64, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.; Alonso-Alonso, R.; Tomás-Roca, L.; Rodríguez-Pinilla, S.M.; Manso-Alonso, R.; Cereceda, L.; Borregón, J.; Villaescusa, T.; Córdoba, R.; Sánchez-Beato, M.; et al. Peripheral T-cell lymphoma: Molecular profiling recognizes subclasses and identifies prognostic markers. Blood Adv. 2021, 5, 5588–5598. [Google Scholar] [CrossRef]
- Dobson, R.; Du, P.Y.; Rásó-Barnett, L.; Yao, W.Q.; Chen, Z.; Casa, C.; Ei-Daly, H.; Farkas, L.; Soilleux, E.; Wright, P.; et al. Early detection of T-cell lymphoma with T follicular helper phenotype by RHOA mutation analysis. Haematologica 2022, 107, 489–499. [Google Scholar] [CrossRef]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsöz-Linke, E.; Müller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2R172 mutations. Mod. Pathol. 2019, 32, 1123–1134. [Google Scholar] [CrossRef]
- Basha, B.M.; Bryant, S.C.; Rech, K.L.; Feldman, A.L.; Vrana, J.A.; Shi, M.; Reed, K.A.; King, R.L. Application of a 5 Marker Panel to the Routine Diagnosis of Peripheral T-Cell Lymphoma With T-Follicular Helper Phenotype. Am. J. Surg. Pathol. 2019, 43, 1282–1290. [Google Scholar] [CrossRef]
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, N.T.; Stein, H.; Gatter, K.C.; Wainscoat, J.S.; Crick, J.; Al Saati, T.; Falini, B.; Delsol, G.; Mason, D.Y. Genotypic analysis of large cell lymphomas which express the Ki-1 antigen. Histopathology 1987, 11, 733–740. [Google Scholar] [CrossRef]
- Foss, H.D.; Anagnostopoulos, I.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef]
- Falini, B.; Bigerna, B.; Fizzotti, M.; Pulford, K.; Pileri, S.A.; Delsol, G.; Carbone, A.; Paulli, M.; Magrini, U.; Menestrina, F.; et al. ALK expression defines a distinct group of T/null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am. J. Pathol. 1998, 153, 875–886. [Google Scholar] [CrossRef]
- Falini, B.; Pulford, K.; Pucciarini, A.; Carbone, A.; De Wolf-Peeters, C.; Cordell, J.; Fizzotti, M.; Santucci, A.; Pelicci, P.G.; Pileri, S.; et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94, 3509–3515. [Google Scholar]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- King, R.L.; Dao, L.N.; McPhail, E.D.; Jaffe, E.S.; Said, J.; Swerdlow, S.H.; Sattler, C.A.; Ketterling, R.P.; Sidhu, J.S.; Hsi, E.D.; et al. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas with DUSP22 Rearrangements. Am. J. Surg. Pathol. 2016, 40, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zettl, A.; Rüdiger, T.; Konrad, M.A.; Chott, A.; Simonitsch-Klupp, I.; Sonnen, R.; Müller-Hermelink, H.K.; Ott, G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am. J. Pathol. 2004, 164, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizotinib-children-and-young-adults-relapsed-or-refractory-systemic-anaplastic-large (accessed on 9 April 2023).
- Piva, R.; Agnelli, L.; Pellegrino, E.; Todoerti, K.; Grosso, V.; Tamagno, I.; Fornari, A.; Martinoglio, B.; Medico, E.; Zamò, A.; et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J. Clin. Oncol. 2010, 28, 1583–1590. [Google Scholar] [CrossRef]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Kucuk, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkaer, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017, 130, 2709–2717. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Trümper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol. 2022, 33, 288–298. [Google Scholar] [CrossRef]
- Miranda, R.N.; Aladily, T.N.; Prince, H.M.; Kanagal-Shamanna, R.; de Jong, D.; Fayad, L.E.; Amin, M.B.; Haideri, N.; Bhagat, G.; Brooks, G.S.; et al. Breast implant-associated anaplastic large-cell lymphoma: Long-term follow-up of 60 patients. J. Clin. Oncol. 2014, 32, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrufino-Schmidt, M.C.; Medeiros, L.J.; Liu, H.; Clemens, M.W.; Hunt, K.K.; Laurent, C.; Lofts, J.; Amin, M.B.; Ming Chai, S.; Morine, A.; et al. Clinicopathologic Features and Prognostic Impact of Lymph Node Involvement in Patients with Breast Implant-associated Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2018, 42, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Medeiros, L.J.; Clemens, M.W.; Ferrufino-Schmidt, M.C.; Pina-Oviedo, S.; Miranda, R.N. Breast implant-associated anaplastic large cell lymphoma: A review. Mod. Pathol. 2019, 32, 166–188. [Google Scholar] [CrossRef]
- Medeiros, L.J.; Marques-Piubelli, M.L.; Sangiorgio, V.F.I.; Ruiz-Cordero, R.; Vega, F.; Feldman, A.L.; Chapman, J.R.; Clemens, M.W.; Hunt, K.K.; Evans, M.G.; et al. Epstein-Barr-virus-positive large B-cell lymphoma associated with breast implants: An analysis of eight patients suggesting a possible pathogenetic relationship. Mod. Pathol. 2021, 34, 2154–2167. [Google Scholar] [CrossRef] [PubMed]
- Mescam, L.; Camus, V.; Schiano, J.M.; Adélaïde, J.; Picquenot, J.M.; Guille, A.; Bannier, M.; Ruminy, P.; Viailly, P.J.; Jardin, F.; et al. EBV+ diffuse large B-cell lymphoma associated with chronic inflammation expands the spectrum of breast implant-related lymphomas. Blood 2020, 135, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Yamashita, D.; Nakamura, S. Nodal EBV+ cytotoxic T-cell lymphoma: A literature review based on the 2017 WHO classification. J. Clin. Exp. Hematop. 2020, 60, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Heavican, T.B.; Bouska, A.; Yu, J.; Lone, W.; Amador, C.; Gong, Q.; Zhang, W.; Li, Y.; Dave, B.J.; Nairismägi, M.L.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef] [Green Version]
- Amador, C.; Greiner, T.C.; Heavican, T.B.; Smith, L.M.; Galvis, K.T.; Lone, W.; Bouska, A.; D’Amore, F.; Pedersen, M.B.; Pileri, S.; et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood 2019, 134, 2159–2170. [Google Scholar] [CrossRef]
- Scott, A.J.; Tokaz, M.C.; Jacobs, M.F.; Chinnaiyan, A.M.; Phillips, T.J.; Wilcox, R.A. Germline variants discovered in lymphoma patients undergoing tumor profiling: A case series. Fam. Cancer 2021, 20, 61–65. [Google Scholar] [CrossRef]
- Leeksma, O.C.; de Miranda, N.F.; Veelken, H. Germline mutations predisposing to diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e532. [Google Scholar] [CrossRef] [Green Version]
- Rendleman, J.; Antipin, Y.; Reva, B.; Adaniel, C.; Przybylo, J.A.; Dutra-Clarke, A.; Hansen, N.; Heguy, A.; Huberman, K.; Borsu, L.; et al. Genetic variation in DNA repair pathways and risk of non-Hodgkin’s lymphoma. PLoS ONE 2014, 9, e101685. [Google Scholar] [CrossRef]
- Usui, Y.; Iwasaki, Y.; Matsuo, K.; Endo, M.; Kamatani, Y.; Hirata, M.; Sugano, K.; Yoshida, T.; Matsuda, K.; Murakami, Y.; et al. Association between germline pathogenic variants in cancer-predisposing genes and lymphoma risk. Cancer Sci. 2022, 113, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Riaz, I.B.; Faridi, W.; Patnaik, M.M.; Abraham, R.S. A Systematic Review on Predisposition to Lymphoid (B and T cell) Neoplasias in Patients with Primary Immunodeficiencies and Immune Dysregulatory Disorders (Inborn Errors of Immunity). Front. Immunol. 2019, 10, 777. [Google Scholar] [CrossRef]



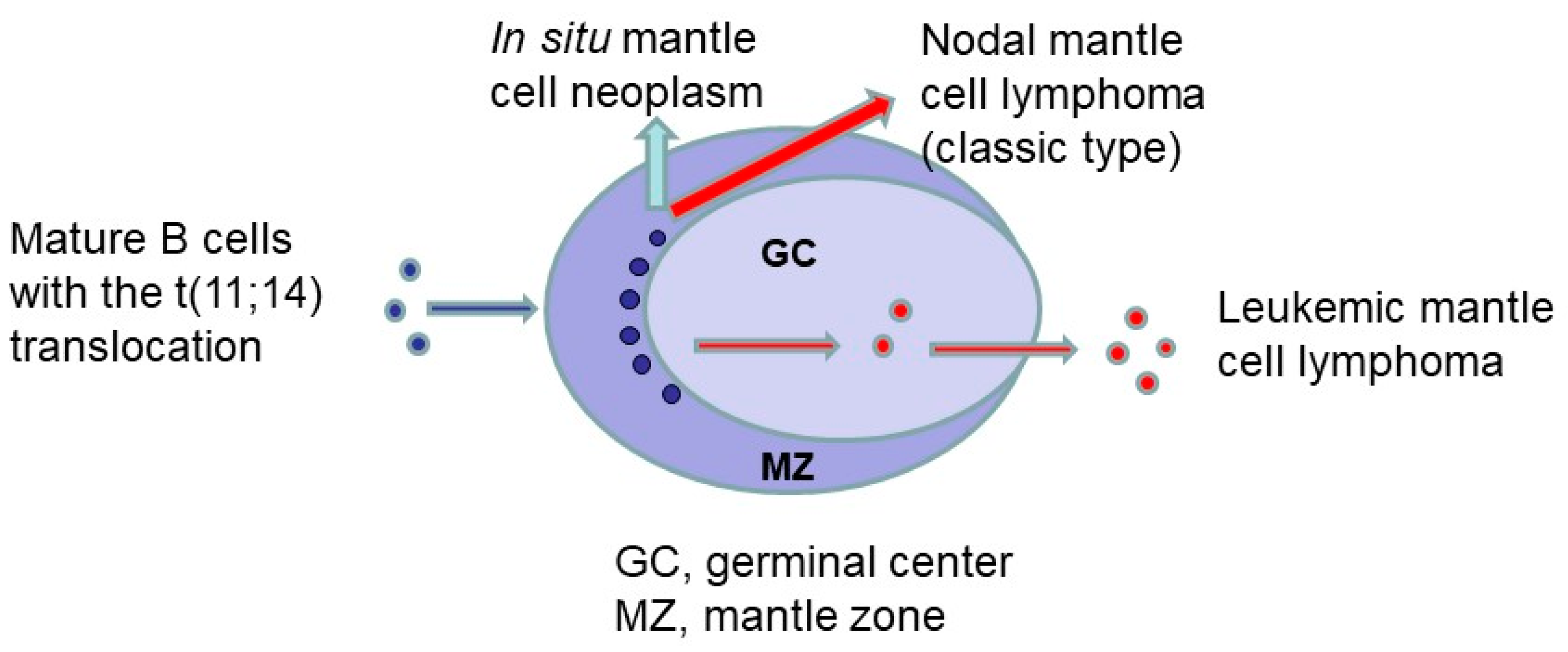{kind=link}
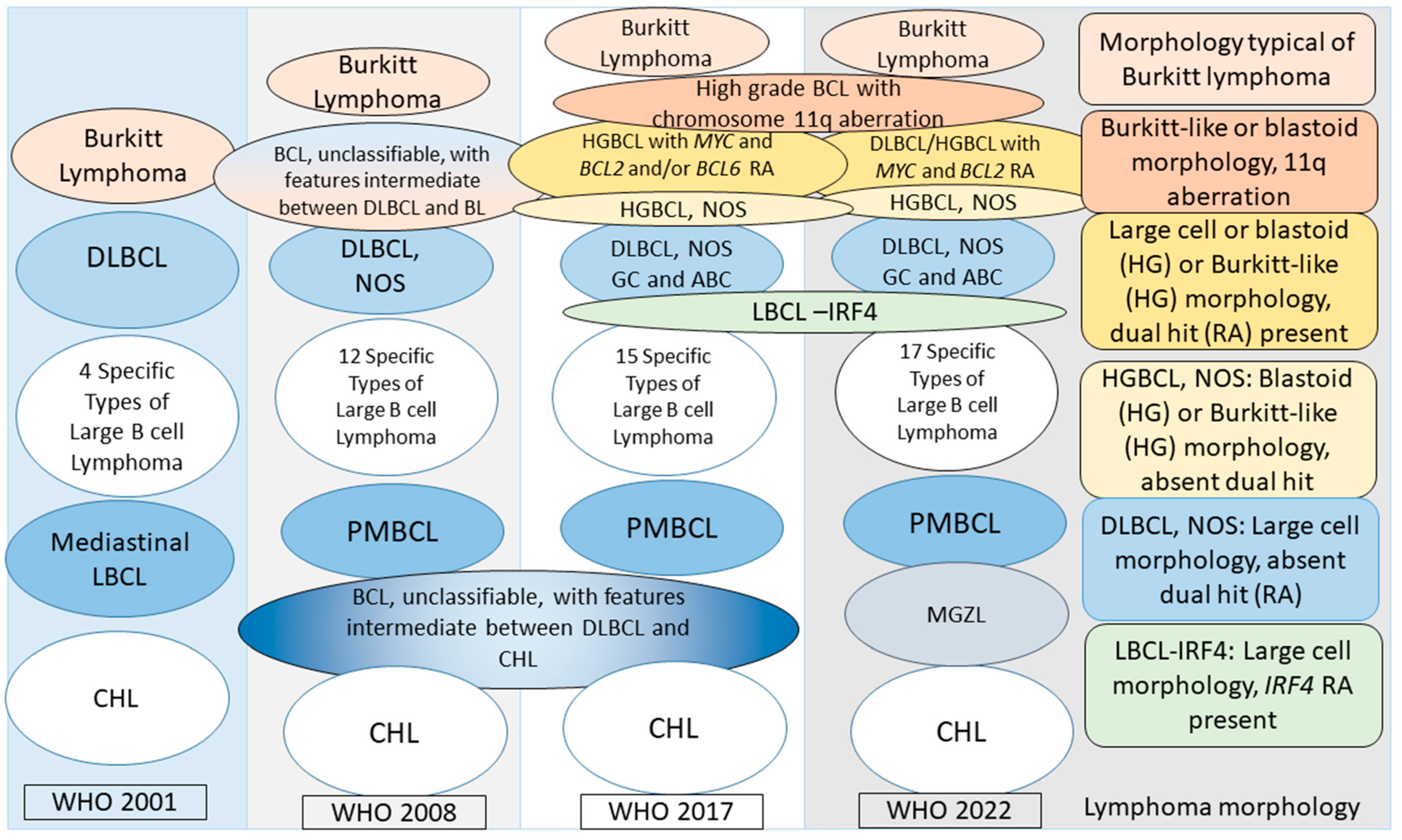{kind=link}
| Cancer Type | Worldwide Incidence | Worldwide Mortality | ||||||
|---|---|---|---|---|---|---|---|---|
| New Cases, N | % of Total N | ASR in Males | ASR in Females | New Deaths, N | % of Total N | ASR in Males | ASR in Females | |
| Non-Hodgkin lymphoma | 544,352 | 2.8% | 6.9 | 4.8 | 259,793 | 2.6% | 3.3 | 2.1 |
| Multiple myeloma | 176,404 | 0.9% | 2.2 | 1.5 | 117,077 | 1.2% | 1.4 | 0.9 |
| Hodgkin lymphoma | 83,087 | 0.4% | 1.2 | 0.8 | 23,376 | 0.2% | 0.3 | 0.2 |
| Leukemia | 474,519 | 2.5% | 6.3 | 4.5 | 311,594 | 3.1% | 4.0 | 2.7 |
| All cancer cases | 19,292,789 | 100% | 222.0 | 186.0 | 9,958,133 | 100% | 120.8 | 84.2 |
| Type of Lymphoma | Age-Adjusted Incidence Rate per 100,000 Persons | |||
|---|---|---|---|---|
| 1992–2001 [5] a | 2000–2018, Blum et al. 2018 [6] b | 2016–2020 SEER [7] | ||
| 15–39 y Age | >39 y Age | |||
| Lymphoid neoplasms, total | 33.65 | |||
| Hodgkin lymphoma | 2.67 | 2.5 | ||
| Classical Hodgkin lymphoma | 2.59 | 3.44 | 2.82 | |
| Nodular sclerosis | 1.63 | |||
| Mixed cellularity/lymphocyte deleted | 0.53 | |||
| Classical Hodgkin lymphoma, NOS | 0.43 | |||
| Nodular lymphocyte predominant HL | 0.08 | 0.14 | 0.17 | |
| Non-Hodgkin lymphomas | 18.7 | |||
| B-cell lymphomas, total | 26.13 | |||
| Precursor B-cell lymphoma | 0.76 | |||
| Precursor B-cell lymphoma, NOS | 0.71 | 0.79 | ||
| Precursor B-cell lymphoma, genetic types | 0.025 | 0.038 | ||
| Mature B-cell lymphoma | ||||
| Chronic lymphocytic leukemia (CLL)/SLL | 5.17 | 0.15 | 13.19 | 4.4 |
| B-cell prolymphocytic leukemia | 0.07 | 0.002 | 0.056 | |
| Mantle cell lymphoma | 0.51 | 0.021 | 1.752 | |
| Lymphoplasmacytic lymphoma/WM | 0.018 | 1.47 | ||
| Lymphoplasmacytic lymphoma | 0.27 | |||
| Waldenström macroglobulinemia | 0.35 | |||
| Follicular lymphoma | 3.18 | 0.5 | 7.78 | 2.5 |
| Marginal zone lymphoma (MZL) | 0.97 | 0.24 | 4.10 | |
| Hairy cell leukemia | 0.33 | 0.075 | 0.62 | |
| Hairy cell variant | 0.004 | 0.31 | ||
| Diffuse large B-cell lymphoma (DLBCL) | 7.14 | 5.5 | ||
| Mediastinal large B-cell lymphoma | 0.079 | 0.041 | ||
| Intravascular large B-cell lymphoma | NA | 0.008 | ||
| Primary effusion lymphoma | 0.006 | 0.02 | ||
| Diffuse large B-cell lymphoma, NOS | 1.45 | 13.7 | ||
| Primary DLBCL of the CNS | 0.058 | 0.578 | ||
| ALK+ Large B-cell lymphoma | 0.002 | 0.002 | ||
| Plasmablastic lymphoma | 0.013 | 0.048 | ||
| Burkitt lymphoma/leukemia | 0.30 | 0.26 | 0.49 | |
| T-cell lymphomas, total | 1.79 | |||
| Precursor T-cell lymphoma | 0.22 | 0.28 | 0.15 | |
| Mature T-cell lymphoma | ||||
| Peripheral T-cell lymphoma | 0.60 | 0.37 | 2.04 | |
| Peripheral T-cell lymphoma, NOS | 0.30 | |||
| Mycosis fungoides/Sezary syndrome | 0.52 | 0.17 | 0.86 | |
| Angioimmunoblastic T-cell lymphoma | 0.05 | |||
| Anaplastic large-cell lymphoma, T/null cell | 0.25 | |||
| Primary cutaneous CD30+ LPDs | 0.036 | 0.17 | ||
| NK/T-cell lymphoma, nasal type | 0.047 | 0.107 | ||
| Type of Lymphoma | 5-y Relative Survival % Based on Patient Ages, 2004–2010 SEER Data [10] | 5-y Relative Survival %Blum et al. 2018 [6] a | 2013–2019 SEER [7] | ||||
|---|---|---|---|---|---|---|---|
| All Ages | 0–19 y | 20–64 y | 65+ y | 15–39 y | >39 y | ||
| Overall | - | - | - | - | 83.10% | 65.40% | |
| Lymphoid neoplasm | 68.1 | 89.0 | 75.8 | 57.4 | - | - | |
| Hodgkin lymphoma | 85.3 | 96.4 | 89.4 | 52.8 | 93.6% | 73.9% | 88.9% |
| Classical Hodgkin lymphoma | - | - | - | - | 94% | 72.70% | |
| Lymphocyte predominant Hodgkin lymphoma | - | - | - | - | 97% | 93% | |
| Non-Hodgkin lymphoma | 66.9 | 87.0 | 74.0 | 58.1 | 74.3% | ||
| B-cell lymphoma | 67.4 | 88.0 | 74.9 | 58.8 | 76.40% | 65.40% | |
| Precursor B-cell lymphoma | 67.6 | 88.0 | 35.4 | 14.0 | |||
| Precursor B-cell lymphoma, NOS | - | - | - | - | 54% | 25% | |
| Precursor B-cell lymphoma, genetic types | - | - | - | - | - | - | |
| Mature B-cell lymphoma | 67.3 | 88.2 | 76.3 | 59.0 | |||
| Chronic lymphocytic leukemia (CLL)/SLL | 79.2 | - | 88.1 | 73.4 | 87% | 80% | 87.7% |
| B-cell prolymphocytic leukemia | 41.6 | - | 51.2 | 36.1 | 66% | 46% | |
| Mantle cell lymphoma | 55.6 | - | 67.5 | 46.0 | 84% | 55% | |
| Lymphoplasmacytic lymphoma/WM | 78.2 | - | 85.1 | 74.1 | 83% | 79% | |
| Lymphoplasmacytic lymphoma | 74.0 | - | 80.7 | 70.1 | - | - | |
| Waldenström macroglobulinemia | 81.5 | - | 88.3 | 77.3 | - | - | |
| Follicular lymphoma | 86.1 | 91.8 | 90.6 | 79.9 | 93% | 85% | 90.6% |
| Marginal zone lymphoma (MZL) | 90.0 | 98.2 | 92.9 | 87.4 | 97% | 88% | |
| Splenic marginal zone lymphoma | 84.8 | - | 87.7 | 81.8 | - | - | |
| Extranodal marginal zone lymphoma | 94.0 | 97.4 | 95.7 | 92.2 | - | - | |
| Nodal marginal zone lymphoma | 83.5 | - | 87.8 | 79.9 | - | - | |
| Hairy cell leukemia | 93.6 | - | 97.6 | 82.5 | 99% | 93% | |
| Hairy cell leukemia variant | 76% | 77% | |||||
| Diffuse large B-cell lymphoma (DLBCL) | 60.5 | 86.4 | 69.9 | 50.7 | 64.7% | ||
| Primary mediastinal large B-cell lymphoma | 81.8 | - | 86.4 | - | 89% | 80% | |
| Intravascular large B-cell lymphoma | 54.8 | - | - | - | - | 2-y: 42% | |
| Primary effusion lymphoma | 22.5 | - | 24.8 | - | 17% | 29% | |
| Diffuse large B-cell lymphoma, NOS | 60.3 | 87.3 | 69.8 | 50.7 | 79% | 59% | |
| Primary DLBCL of the CNS | - | - | - | - | 45% | 26% | |
| ALK+ Large B-cell lymphoma | - | - | - | - | 2-y 66.9% | 2-y 100% | |
| Plasmablastic lymphoma | 2-y 50.90% | 2-y 45% | |||||
| Burkitt lymphoma/leukemia | 58.3 | 88.6 | 55.8 | 31.0 | 68% | 45% | |
| Non-Hodgkin lymphoma, T and NK | - | - | - | - | 69.20% | 57.5% | |
| T-cell lymphoma | 63.1 | 83.3 | 66.3 | 49.6 | |||
| Precursor T-cell lymphoma | 62.4 | 84.5 | 44.8 | 8.0 | 59% | 32% | |
| Mature T-cell lymphoma | 63.4 | 82.7 | 68.1 | 50.7 | |||
| Peripheral T-cell lymphoma | 56.0 | 81.6 | 62.4 | 43.2 | 68% | 48.3% | |
| Peripheral T-cell lymphoma, NOS | 37.0 | 60.4 | 42.5 | 29.1 | |||
| Mycosis fungoides/Sezary syndrome | 89.4 | 94.9 | 93.0 | 80.7 | 96.20% | 88.2% | |
| Mycosis fungoides | 90.6 | 94.9 | 93.6 | 83.0 | - | - | |
| Sezary syndrome | 44.1 | - | 57.1 | 33.3 | - | - | |
| Angioimmunoblastic T-cell lymphoma | 43.4 | - | 52.6 | 35.2 | - | - | |
| Anaplastic large-cell lymphoma, T/null cell | 58.1 | 85.2 | 62.0 | 32.0 | - | - | |
| ALCL, primary cutaneous | 90.9 | - | 94.5 | 81.3 | - | - | |
| Primary cutaneous CD30+ LPDs | - | - | - | - | 94% | 87.3% | |
| NK/T-cell lymphoma, nasal type | 41.6 | - | 39.8 | 43.5 | 43.30% | 37.7% | |
| Fifth Edition WHO classification 2022 [11,12] | International Consensus Classification 2022 [13] |
|---|---|
| B-cell lymphoid proliferations and lymphomas * 1. Tumor-like lesions with B-cell predominance Reactive B-cell rich lymphoid proliferations that can mimic lymphoma IgG4-related disease Unicentric Castleman disease Idiopathic multicentric Castleman disease KSHV/HHV8-associated multicentric Castleman disease 2. Mature B-cell neoplasms Pre-neoplastic and neoplastic small lymphocytic proliferations Monoclonal B-cell lymphocytosis Chronic lymphocytic leukemia/small lymphocytic lymphoma Splenic B-cell lymphomas and leukemias Hairy cell leukemia Splenic marginal zone lymphoma Splenic diffuse red pulp small B-cell lymphoma Splenic B-cell lymphoma/leukemia with prominent nucleoli Lymphoplasmacytic lymphoma Lymphoplasmacytic lymphoma Marginal zone lymphoma Extranodal marginal zone lymphoma of MALT Primary cutaneous marginal zone lymphomaa Nodal marginal zone lymphoma Pediatric nodal marginal zone lymphoma Follicular lymphoma In situ follicular B-cell neoplasm Follicular lymphoma Classic follicular lymphoma Follicular lymphoma with uncommon features Blastoid or large centrocyte variant by cytology Prediminantly diffuse pattern Pediatric-type follicular lymphoma Duodenal-type follicular lymphoma Primary cutaneous follicle center lymphoma Mantle cell lymphoma In situ mantle cell neoplasm Mantle cell lymphoma Leukemic non-nodal mantle cell lymphoma Transformations of indolent B-cell lymphomasa Transformations of indolent B-cell lymphomas Large B-cell lymphomas Diffuse large B-cell lymphoma (DLBCL), NOS T-cell/histiocyte-rich large B-cell lymphoma DLBCL/high-grade B-cell lymphoma with MYC and BCL2 rearrangements ALK-positive large B-cell lymphoma Large B-cell lymphoma with IRF4 rearrangement High-grade B-cell lymphoma with 11q chromosomal aberrations Lymphomatoid granulomatosis Epstein–Barr virus (EBV)+ DLBCL DLBCL associated with chronic inflammation Fibrin-associated large B-cell lymphoma a Fluid overload-associated large B-cell lymphomaa Plasmablastic lymphoma Primary large B-cell lymphoma of immune-privileged sites a Primary diffuse large B-cell lymphoma of the CNS Primary large B-cell lymphoma of the vitreoretinal areas a Primary large B-cell lymphoma of the testis a Primary cutaneous diffuse large B-cell lymphoma, leg type Intravascular large B-cell lymphoma Primary mediastinal large B-cell lymphoma Mediastinal grey zone lymphoma High-grade B-cell lymphoma, NOS Burkitt lymphoma Burkitt lymphoma KSHV/HHV8-associated B-cell lymphoid proliferations and lymphomas Primary effusion lymphoma KSHV/HHV8+ DLBCL KSHV/HHV8+ germinotropic lymphoproliferative disorder Lymphoid proliferations and lymphomas associated with immune deficiency and dysregulation a Hyperplasias arising in immune deficiency/dysregulationa Polymorphic LPDs arising in immune deficiency/dysregulation a Epstein–Barr virus (EBV)+ mucocutaneous ulcer Lymphomas arising in immune deficiency/dysregulationa Inborn error of immunity-associated lymphoid proliferations and lymphomas 3. Hodgkin lymphoma Classic Hodgkin lymphoma Nodular lymphocyte predominant Hodgkin lymphoma 4. Plasma cell neoplasms and other diseases with paraproteins Monoclonal gammopathies Cold agglutinin disease a IgM MGUS Non-IgM MGUS Monoclonal gammopathy of renal significance a Diseases with monoclonal immunoglobulin deposition Immunoglobulin-related (AL) amyloidosis Monoclonal immunoglobulin deposition disease Heavy chain diseases Mu heavy chain disease Gamma heavy chain disease Alpha heavy chain disease Plasma cell neoplasms Plasmacytoma Plasma cell myeloma/multiple myeloma Plasma cell neoplasms with associated paraneoplastic syndrome POEMS syndrome TEMPI syndrome AESOP syndrome a | Mature B-cell neoplasms Chronic lymphocytic leukemia/small lymphocytic lymphoma Monoclonal B-cell lymphocytosis CLL type Non-CLL type B-cell prolymphocytic leukemia Splenic marginal zone lymphoma Hairy cell leukemia Splenic B-cell lymphoma/leukemia, unclassifiable (provisional) Splenic diffuse red pulp small B-cell lymphoma (provisional) Hairy cell leukemia-variant (provisional) Lymphoplasmacytic lymphoma Waldenström macroglobulinemia IgM monoclonal gammopathy of undetermined significance IgM MGUS, plasma cell type a IgM MGUS, NOS a Primary cold agglutinin disease a Heavy chain diseases Mu heavy chain disease Gamma heavy chain disease Alpha heavy chain disease Plasma cell neoplasms Non-IgM MGUS Multiple myeloma (Plasma cell myeloma) a Multiple myeloma NOS Multiple myeloma with recurrent genetic abnormality Multiple myeloma with CCND family translocation Multiple myeloma with MAF family translocation Multiple myeloma with NSD2 translocation Multiple myeloma with hyperdiploidy Solitary plasmacytoma of bone Extraosseous plasmacytoma Monoclonal immunoglobulin deposition diseases Immunoglobulin light chain amyloidosis (AL) a Localized AL amyloidosisa Light chain and heavy chain deposition disease Extranodal marginal zone lymphoma of MALT Primary cutaneous marginal zone lymphoproliferative disorder a Nodal marginal zone lymphoma Pediatric nodal marginal zone lymphoma (provisional) Follicular lymphoma In situ follicular neoplasia Duodenal-type follicular lymphoma BCL2-R negative, CD23+ follicle center lymphoma (provisional) Primary cutaneous follicle center lymphoma Pediatric-type follicular lymphoma Testicular follicular lymphoma a Large B-cell lymphoma with IRF4 rearrangement a Mantle cell lymphoma In situ mantle cell neoplasia Leukemic non-nodal mantle cell lymphoma Diffuse large B-cell lymphoma (DLBCL), NOS Germinal center B-cell subtype Activated B-cell subtype Large B-cell lymphoma with 11q aberration (provisional) a Nodular lymphocyte predominant B-cell lymphoma a T-cell/histiocyte-rich large B-cell lymphoma Primary DLBCL of the central nervous system Primary DLBCL of the testisa Primary cutaneous DLBCL, leg type Intravascular large B-cell lymphoma HHV8 and EBV-negative primary effusion-based lymphoma * (provisional) EBV+ mucocutaneous ulcer a EBV+ DLBCL, NOS DLBCL associated with chronic inflammation Fibrin-associated DLBCL Lymphomatoid granulomatosis EBV+ polymorphic B-cell lymphoproliferative disorder, NOS a ALK+ large B-cell lymphoma Plasmablastic lymphoma HHV8-associated lymphoproliferative disorders Multicentric Castleman disease HHV8+ germinotropic lymphoproliferative disorder HHV8+ DLBCL, NOS Primary effusion lymphoma Burkitt lymphoma High-grade B-cell lymphoma with MYC and BCL2 rearrangements a High-grade B-cell lymphoma with MYC and BCL6 rearrangements (provisional) a High-grade B-cell lymphoma, NOS Primary mediastinal large B-cell lymphoma Mediastinal gray-zone lymphoma a Classic Hodgkin lymphoma Nodular sclerosis classic Hodgkin lymphoma Lymphocyte-rich classic Hodgkin lymphoma Mixed cellularity classic Hodgkin lymphoma Lymphocyte-depleted classic Hodgkin lymphoma Immunodeficiency-associated lymphoproliferative disorders Post-transplant lymphoproliferative disorders (PTLDs) Non-destructive PTLDs Plasmacytic hyperplasia PTLD Infectious mononucleosis PTLD Florid follicular hyperplasia PTLD Polymorphic PTLD Monomorphic PTLD (B-cell and T-cell/NK-cell types) b Classic Hodgkin lymphoma PTLD b Other iatrogenic immunodeficiency-associated LPDs |
| Patient Age | Female | Male | ||||||
|---|---|---|---|---|---|---|---|---|
| Incidence Rate per 100,000 Individuals during 2000–2020, Year(s) at Diagnosis | Trend 2000–2019 | Incidence Rate per 100,000 Individuals during 2000–2020, Year(s) at Diagnosis | Trend 2000–2019 | |||||
| 2000 to 2020 | Maximum | Lowest | 2000 to 2020 | Maximum | Lowest | |||
| Hodgkin Lymphomas | ||||||||
| All ages | 2.4 (2000) to 2.2 (2020) | 2.6 (2008, 2006) | 2.1 (2018) | Falling | 3.2 (2000) to 2.8 (2020) | 3.4 (2005) | 2.8 (2020, 2017) | Falling |
| <15 years | 0.5 (2000) to 0.4 (2020) | 0.6 (2008) | 0.4 (2020, and other) | NS | 0.7 (2000) to 0.7 (2020) | 0.8 (2005, 2006) | 0.6 (2010) | NS |
| 15–39 years | 3.7 (2000) to 3.5 (2020) | 3.9 (2005, 2006) | 3.2 (2018) | Falling | 3.9 (2000) to 3.3 (2020) | 4.0 (2006, 2007) | 3.3 (2020, 2017) | Falling |
| 40–64 years | 2.0 (2000) to 1.8 (2020) | 2.1 (2019, and other) | 1.8 (2020, 2018) | Falling | 3.5 (2000) to 2.7 (2020) | 3.6 (2001, 2006) | 2.7 (2020) | Falling |
| 65–74 years | 2.9 (2000) to 2.2 (2020) | 3.5 (2004) | 2.2 (2020) | Falling | 4,7 (2000) to 3.8 (2020) | 5.5 (2005) | 3.8 (2020) | Falling |
| 75+ years | 3.2 (2000) to 2.1 (2020) | 4.6 (2006) | 2.7 (2015) | Falling | 4.4 (2000) to 5.7 (2020) | 6.5 (2010) | 4.3 (2001) | See note a |
| Non-Hodgkin Lymphomas | ||||||||
| All ages | 16.4 (2000) to 14.8 (2020) | 17.0 (2003, 2004) | 14.8 (2020) | See note b | 23.0 (2000) to 21.5 (2020) | 24.7 (2008, 2005) | 21.5 (2020) | See note b |
| <15 years | 0.6 (2000) to 0.6 (2020) | 0.8 (2016, and other) | 0.5 (2009) | Rising | 1.1 (2000) to 1.4 (2020) | 1.6 (2014) | 1.1 (2003, 2000) | See note c |
| 15–39 years | 3.4 (2000) to 3.4 (2020) | 3.6 (2009) | 3.1 (2011) | NS | 5.3 (2000) to 4.4 (2020) | 5.3 (2000, 2001) | 4.4. (2020) | Falling |
| 40–64 years | 18.4 (2000) to 16.3 (2020) | 19.0 (2003) | 16.3 (2020) | Falling | 26.6 (2000) to 22.1 (2020) | 27.2 (2004) | 22.1 (2020) | See note d |
| 65–74 years | 62.7 (2000) to 54.3 (2020) | 64.4 (2010) | 54.3 (2020) | Falling | 81.3 (2000) to 78.6 (2020) | 90.9 (2007) | 78.6 (2020) | See note e |
| 75+ years | 88.0 (2000) to 81.4 (2020) | 96.4 (2009) | 81.4 (2020) | Falling | 122.8 (2000) to 128.4 (2020) | 145.3 (2009) | 122.8 (2000) | NS |
| The Outcomes of 31 Reported Cases of In Situ Mantle Cell Neoplasia | References |
|---|---|
A proportion of 29% (9/31) of ISMCN cases occurred in a background of a composite lymphoma
| [109,110,111,112,113,114,115] |
In 22 ISMCN patients without a composite lymphoma
| [109,111,116,119] |
| [109,113,116] |
| [109,120] |
| [107] |
| [107,109,121,122] |
| [123] |
| CD19 | CD20 | CD5 | CD43 | CD23 | CD10 | BCL2 | BCL1 | Ig Κ/λ | Other | |
|---|---|---|---|---|---|---|---|---|---|---|
| CLL/SLL | + | + Dim a | + weak b | +/- | +/− | − | + | − c | + Dim a | FMC7−CD81− CD200+ LEF1+ |
| MCL | + | + Bright | + | + | −/+ | − | + | + | + bright | FMC7+SOX11+ |
| FL | + | + | −/rare + | −/rare + | + | +/- | + d | - | + | BCL6+/− |
| MZL e | + | + | −/+weak | −/+ | −/+ | −/rare + | + f | − | + | IgD+ g FMC7+ CD11c +/− CD27+ CD103- |
| HCL | + | + | − | − | −/+ | −/rare + | + | + weak | + bright | Bright CD11c+ CD22+ h,i, CD103+ |
| LPL | + | + | −/+ rare | −/+ j | −/+ rare | −/+ rare | + | − | + k | IgM+ |
| TdT | CD20 | CD10 | BCL2 | BCL6 | MUM1 | Ki-67 | MYC | CD5 | LMO2 | Other | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BL [217,243] | − | + | + | − | + | − | >95% | + | − | − | CD45dim+ CD16/56− CD38bright+ CD44− CD43+ |
| HGBCL-11q [217] | − | + | + | − | + | − | High >90% | + | − | +/− | CD45bright+ CD38 (not bright)+ CD16/56+ CD8+/− CD43+/− CD44−/+ |
| LBCL-IRF4 [212,244] | − | + | +/− | −/+ | + | + | high | −/+ | −/+ [245] | NA | EBV− BLIMP1− |
| DLBCL GC [243] | − | + | + | −/+ | + | − | low | −/+ [243] | −/+ | +/− [243] | FOXP1− cyclin D1− |
| DLBCL ABC | − | + | − | + | −/+ | + | low | −/+ | −/+ | − | FOXP1+ cyclin D1− |
| PCNS-LBCL or PVR-LBCL | − | + | − | + | + | + | High 80–90% | − | − | NA | EBV−; If +, consider IDD |
| PCNS-LBCL or PVR-LBCL in IDD | − | + | − | + | + | + | High 80–90% | − | − | NA | EBV+ |
| IVBCL [239] | − | + | −/+ | + | + | + | high | + | +/− | NA | PD-L1+ |
| Primary Mediastinal Lymphomas According to Anatomic Site | |
|---|---|
| Anterior mediastinum | |
| T lymphoblastic lymphoma | 2–5% of all non-Hodgkin lymphomas |
| Primary mediastinal (thymic) large B-cell lymphoma (PMBCL) | 2–3% of all non-Hodgkin lymphomas |
| Primary thymic marginal zone lymphoma of MALT type | Very rare |
| Mediastinal grey zone lymphoma (MGZL) | Very rare |
| Anterior, middle, or posterior mediastinum | |
| Primary mediastinal (nonthymic) DLBCL | 5–9% of all non-Hodgkin lymphomas |
| T-cell lymphomas | Very rare |
| Peripheral T-cell lymphoma, NOS | |
| Anaplastic large-cell lymphoma (ALCL) | |
| Breast implant-associated anaplastic large-cell lymphoma | |
| Primary mediastinal small B-cell lymphomas | Exceedingly rare |
| Primary mediastinal plasmacytoma | Exceedingly rare |
| Pleura with a mass | |
| Primary pleural DLBCL, NOS, usually non-GCB type | |
| DLBCL associated with chronic inflammation/pyothorax-associated lymphoma | Very rare |
| Pleura and/or pericardium without a mass | |
| Primary effusion lymphoma | Very rare |
| Heart or pericardium with a mass | |
| Primary cardiac DLBCL, NOS, usually non-GCB type | Very rare |
| Heart or pericardium without a mass (incidental microscopic finding) | |
| Fibrin-associated DLBCL, the mass is a myxoma or a thrombus | Exceedingly rare |
| Lymphoma Type | CD45/LCA | CD20 | CD3 | PAX5 | CD30 | CD15 | EBV | EMA | ALK | Other |
|---|---|---|---|---|---|---|---|---|---|---|
| NLPHL | + | + | − | + | − | − | − | +/− | − | CD79a+ OCT2+ BOB1+ |
| CHL | − | −/+, het | −/rare + | + (weak) a | + | +/− | +/− | − | − | OCT2−/+ BOB1−/+ |
| PTCL NOS | + | −/+ rare | + | − | −/+ | − | −/+ | − | − | One TFH marker may be positive in PTCL NOS |
| TFH lymphomas b | + | − | + | − | −/+ | −/+ | + c/− | − | − | CD2+ CD4+ CD5+ CD8- BCL6+ ICOS+ CD10+/- PD1+ CXCL13+; CD21+ FDC proliferation in the paracortex |
| ALCL | +/− | − | often | − | + | − | − | + | +/− | CD2+ CD5+ CD4+/− TIA+ perforin+ granzyme B+ |
| ALK+ LBCL | + | − often | − | − | −/+ rare | −/+ | − d | + | + e | CD138+ MUM1+ |
| PMBCL | + | + | − | + | + het, weak | +/− | − | − | − | CD23+ MAL+ CD200+ or PD-L1, PD-L2+ f |
| Fifth edition WHO Classification 2022 [11,12] | International Consensus Classification 2022 [13] |
|---|---|
| T-cell and NK-cell lymphoid proliferations and lymphomas Tumor-like lesions with T-cell predominance a Kikuchi-Fujimoto disease a Autoimmune lymphoproliferative syndrome a Indolent T lymphoblastic proliferation a Mature T-cell and NK-cell leukemias T-prolymphocytic leukaemia T-large granular lymphocytic leukaemia NK-large granular lymphocytic leukaemia Adult T-cell leukaemia/lymphoma Sezary syndrome Aggressive NK-cell leukaemia Primary cutaneous T-cell lymphoid proliferations and lymphomas Primary cutaneous CD4-positive small or medium T-cell LPD Primary cutaneous acral CD8-positive T-cell LPD Mycosis fungoides Primary cutaneous CD30+ T-cell LPD: Lymphomatoid papulosis Primary cutaneous CD30+ T-cell LPD Primary cutaneous ALCL Subcutaneous panniculitis-like T-cell lymphoma Primary cutaneous gamma/delta T-cell lymphoma Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma Primary cutaneous peripheral T-cell lymphoma, NOS a Intestinal T-cell and NK-cell lymphoid proliferations and lymphomas Indolent T-cell lymphoma of the gastrointestinal tract a Indolent NK-cell LPD of the gastrointestinal tract Enteropathy-associated T-cell lymphoma Monomorphic epitheliotropic intestinal T-cell lymphoma Intestinal T-cell lymphoma, NOS Hepatosplenic T-cell lymphoma Hepatosplenic T-cell lymphoma Anaplastic large-cell lymphoma ALK-positive anaplastic large-cell lymphoma ALK-negative anaplastic large-cell lymphoma Breast implant-associated anaplastic large-cell lymphoma Nodal T-follicular helper (TFH) cell lymphoma a Nodal TFH cell lymphoma, angioimmunoblastic-type Nodal TFH cell lymphoma, follicular-type Nodal TFH cell lymphoma, NOS Other peripheral T-cell lymphomas Peripheral T-cell lymphoma, NOS EBV+ NK-cell and T-cell lymphomas EBV+ nodal T- and NK-cell lymphoma a Extranodal NK/T-cell lymphoma EBV+ T-cell and NK-cell lymphoid proliferations and lymphomas of childhood Severe mosquito bite allergy Hydroa vacciniforme lymphoproliferative disorder Systemic chronic active EBV disease Systemic EBV-positive T-cell lymphoma of childhood | Mature T-cell and NK-cell neoplasms T-cell prolymphocytic leukemia T-cell large granular lymphocytic leukemia Chronic lymphoproliferative disorder of NK-cells (provisional) Adult T-cell leukemia/lymphoma EBV+ T-cell/NK-cell lymphoproliferative disorders of childhood b Hydroa vacciniforme lymphoproliferative disorder Classic Systemic Severe mosquito bite allergy Chronic active EBV disease, systemic (T-cell and NK-cell phenotype) Systemic EBV+ T-cell lymphoma of childhood Extranodal NK/T-cell lymphoma, nasal type Aggressive NK-cell leukemia Primary nodal EBV+ T-cell/NK-cell lymphoma b (provisional) Enteropathy-associated T-cell lymphoma Type II refractory celiac disease b Monomorphic epitheliotropic intestinal T-cell lymphoma Intestinal T-cell lymphoma, NOS Indolent clonal T-cell LPD of the gastrointestinal tract b Indolent NK-cell LPD of the gastrointestinal tract b Hepatosplenic T-cell lymphoma Mycosis fungoides Sézary syndrome Primary cutaneous CD30+ T-cell lymphoproliferative disorders Lymphomatoid papulosis Primary cutaneous anaplastic large-cell lymphoma Primary cutaneous small/medium CD4+ T-cell LPD Subcutaneous panniculitis-like T-cell lymphoma Primary cutaneous gamma-delta T-cell lymphoma Primary cutaneous acral CD8+ T-cell LPD b Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma Peripheral T-cell lymphoma, NOS Follicular helper T-cell lymphoma b Follicular helper T-cell lymphoma, angioimmunoblastic type (angioimmunoblastic T-cell lymphoma) Follicular helper T-cell lymphoma, follicular type Follicular helper T-cell lymphoma, NOS Anaplastic large-cell lymphoma, ALK positive Anaplastic large-cell lymphoma, ALK negative Breast implant-associated anaplastic large-cell lymphoma |
| WHO Classification, Fifth Edition, 2022 [11,12] | WHO Classification, Revised Fourth Edition, 2017 [100] | International Consensus Classification, 2022 [13] |
|---|---|---|
| T-cell and NK-cell neoplasms | ||
| NK-large granular lymphocytic leukemia (LGLL) Cutaneous T-cell lymphomas Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma; Primary cutaneous acral CD8+ T-cell lymphoproliferative disorder (LPD); Primary cutaneous CD4+ small or medium T-cell LPD Indolent T-cell lymphoma of the GI tract Indolent NK-cell LPD of the GI tract Breast implant-associated ALCL Nodal T follicular helper (TFH) cell lymphomas Nodal T-follicular helper (TFH) cell lymphoma, angioimmunoblastic-type Nodal T-follicular helper (TFH) cell lymphoma, follicular-type Nodal T-follicular helper (TFH) cell lymphoma, NOS Hydroa vacciniforme LPD Systemic chronic active EBV disease (not recommended: chronic active EBV disease or infection) EBV+ inflammatory FDC/fibroblastic reticular cell sarcoma | Chronic LPD of NK-cells Cutaneous T-cell lymphomas Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (provisional); Primary cutaneous acral CD8+ T-cell lymphoma (provisional); Primary cutaneous CD4+ small/medium T-cell LPD (provisional) Indolent T-cell LPD of the GI tract (provisional) (Not previously included) Breast implant-associated ALCL (provisional) Nodal lymphomas of TFH origin Angioimmunoblastic T-cell lymphoma Follicular T-cell lymphoma (provisional) Nodal peripheral T-cell lymphoma with TFH phenotype (provisional) Hydroa vacciniforme-like LPD Chronic active EBV infection EBV+ inflammatory FDC/fibroblastic reticular cell sarcoma | Chronic LPD of NK-cells (provisional) Cutaneous T-cell lymphomas Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma; Primary cutaneous acral CD8+ T-cell lymphoproliferative disorder (LPD); Primary cutaneous CD4+ small/medium T-cell LPD Indolent clonal T-cell LPD of the GI tract Indolent NK-cell LPD of the GI tract Breast implant-associated ALCL Follicular helper T-cell (TFH) lymphoma Follicular helper T-cell lymphoma, angioimmunoblastic type Follicular helper T-cell lymphoma, follicular type Follicular helper T-cell lymphoma, NOS Hydroa vacciniforme LPD Chronic active EBV disease EBV+ inflammatory FDC/fibroblastic reticular cell tumor |
| Hodgkin lymphoma | ||
| Nodular lymphocyte predominant Hodgkin lymphoma | Nodular lymphocyte predominant Hodgkin lymphoma | Nodular lymphocyte predominant B-cell lymphoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 3: Mature Leukemias/Lymphomas. Lymphatics 2023, 1, 155-219. https://doi.org/10.3390/lymphatics1020012
Kansal R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 3: Mature Leukemias/Lymphomas. Lymphatics. 2023; 1(2):155-219. https://doi.org/10.3390/lymphatics1020012
Chicago/Turabian StyleKansal, Rina. 2023. "Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 3: Mature Leukemias/Lymphomas" Lymphatics 1, no. 2: 155-219. https://doi.org/10.3390/lymphatics1020012
APA StyleKansal, R. (2023). Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 3: Mature Leukemias/Lymphomas. Lymphatics, 1(2), 155-219. https://doi.org/10.3390/lymphatics1020012