



Biomolecules 2023, 13(3), 527; https://doi.org/10.3390/biom13030527 - 13 Mar 2023
Cited by 4 | Viewed by 3182
Abstract
►
Show Figures
Research in the field of biochemistry and cellular biology has entered a new phase due to the discovery of phase separation driving the formation of biomolecular condensates, or membraneless organelles, in cells. The implications of this novel principle of cellular organization are vast
[...] Read more.
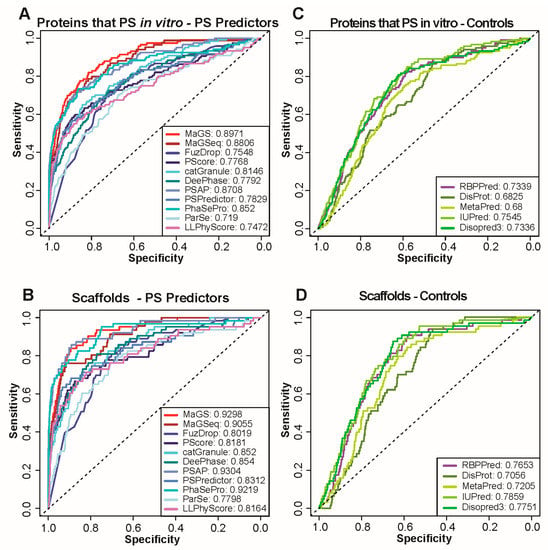
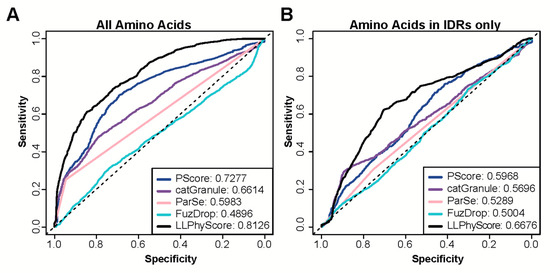
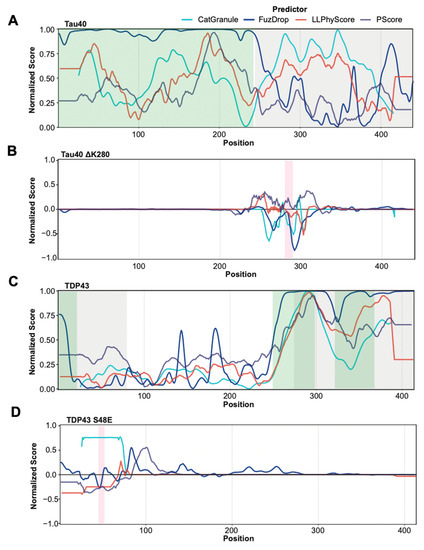
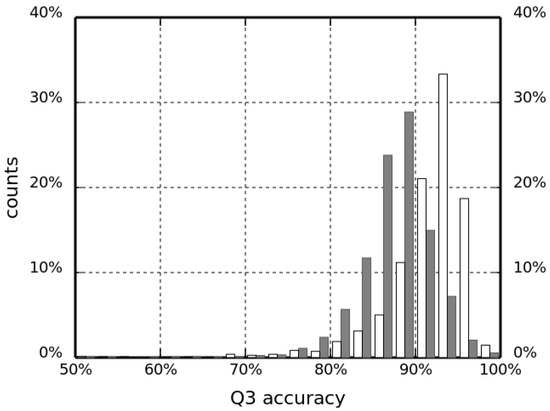
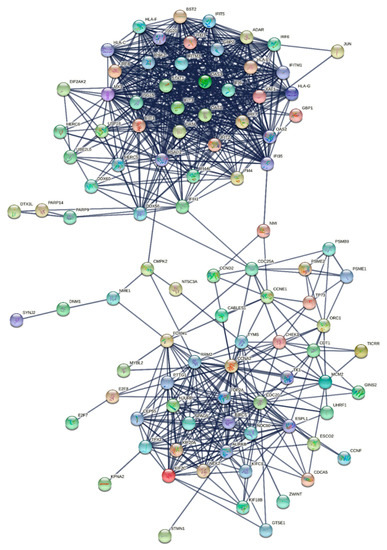
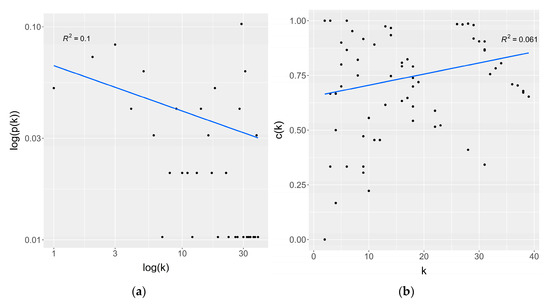
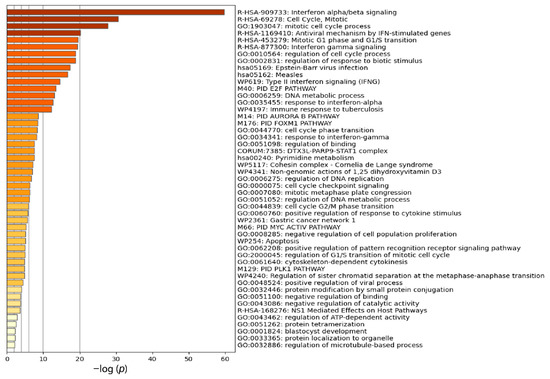
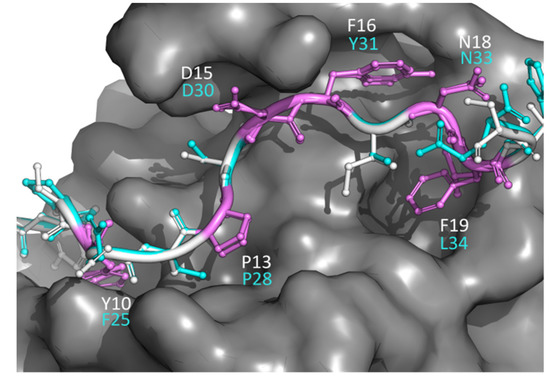
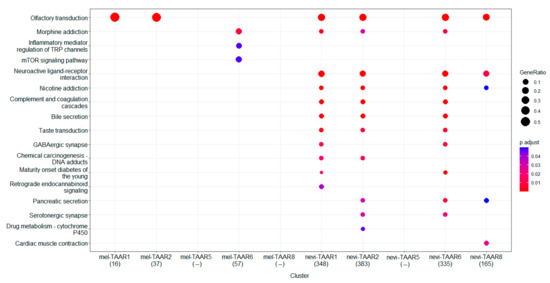
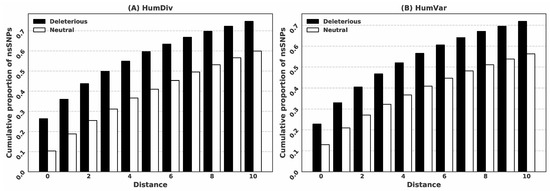
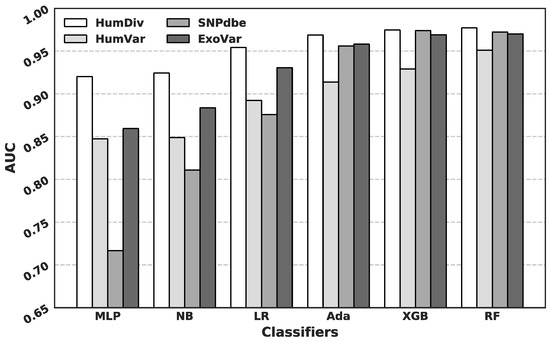
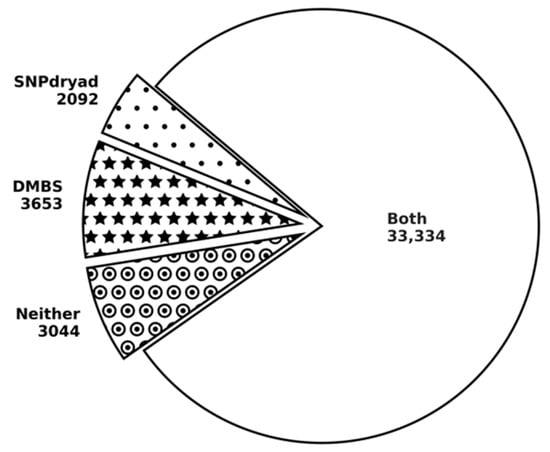
Research in the field of biochemistry and cellular biology has entered a new phase due to the discovery of phase separation driving the formation of biomolecular condensates, or membraneless organelles, in cells. The implications of this novel principle of cellular organization are vast and can be applied at multiple scales, spawning exciting research questions in numerous directions. Of fundamental importance are the molecular mechanisms that underly biomolecular condensate formation within cells and whether insights gained into these mechanisms provide a gateway for accurate predictions of protein phase behavior. Within the last six years, a significant number of predictors for protein phase separation and condensate localization have emerged. Herein, we compare a collection of state-of-the-art predictors on different tasks related to protein phase behavior. We show that the tested methods achieve high AUCs in the identification of biomolecular condensate drivers and scaffolds, as well as in the identification of proteins able to phase separate in vitro. However, our benchmark tests reveal that their performance is poorer when used to predict protein segments that are involved in phase separation or to classify amino acid substitutions as phase-separation-promoting or -inhibiting mutations. Our results suggest that the phenomenological approach used by most predictors is insufficient to fully grasp the complexity of the phenomenon within biological contexts and make reliable predictions related to protein phase behavior at the residue level.
Full article
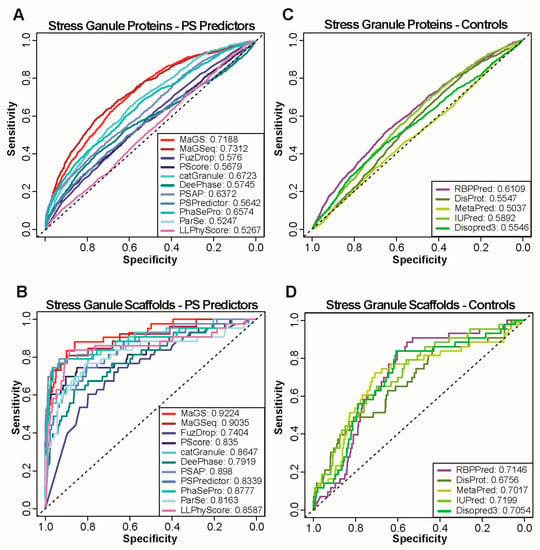
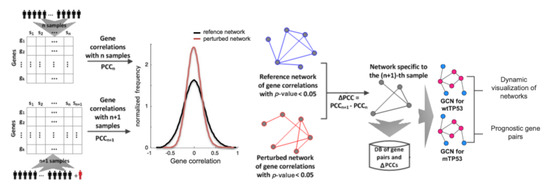
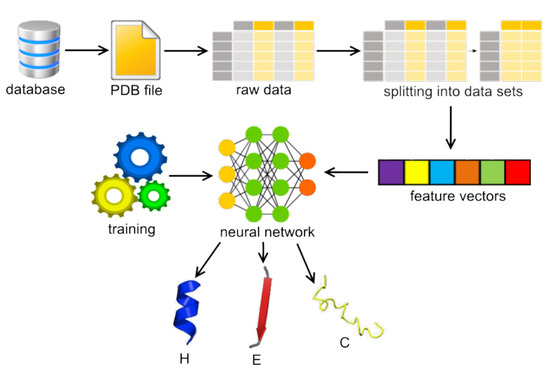
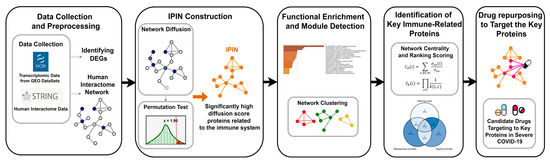
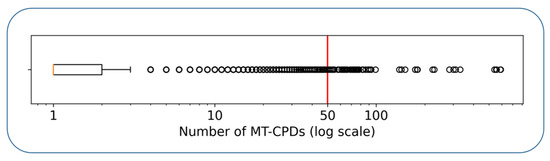
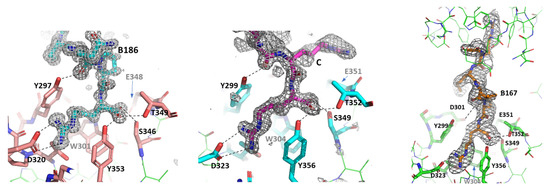
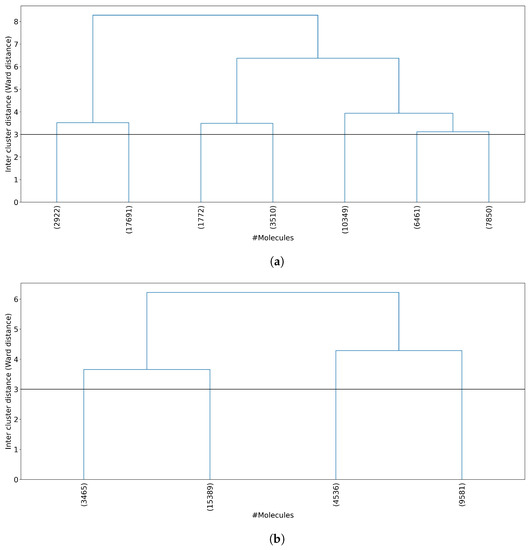
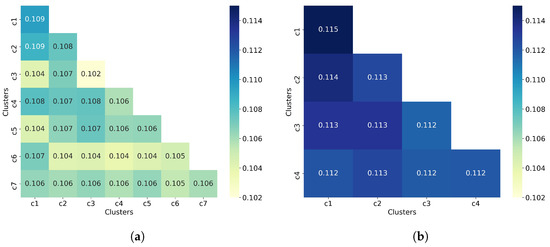
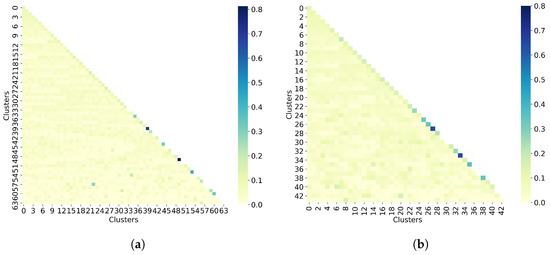
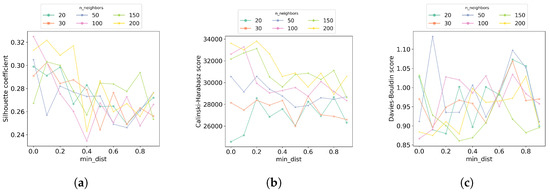
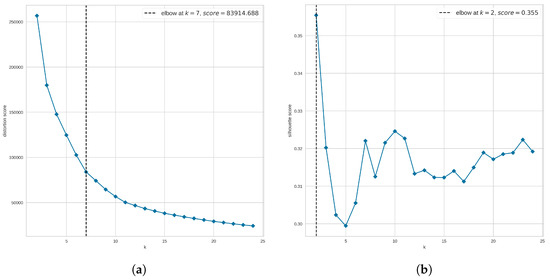
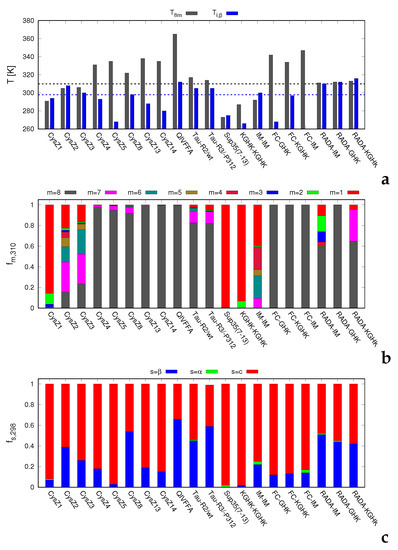
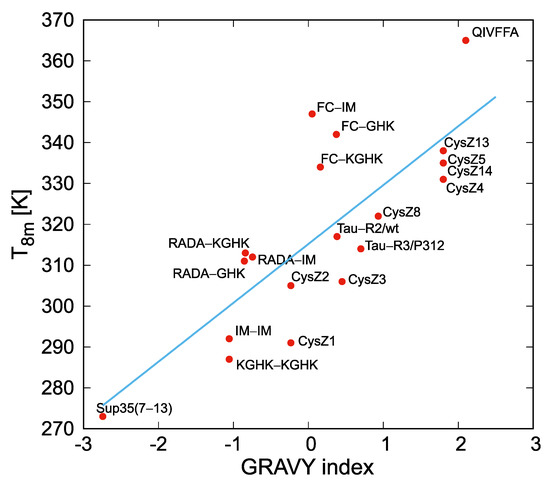
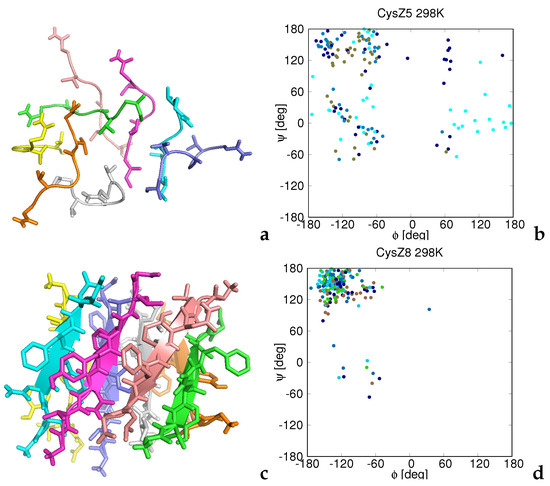
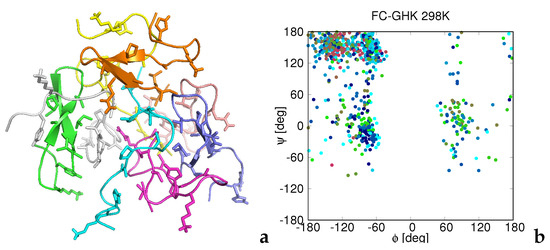
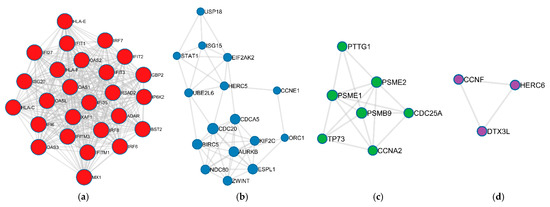
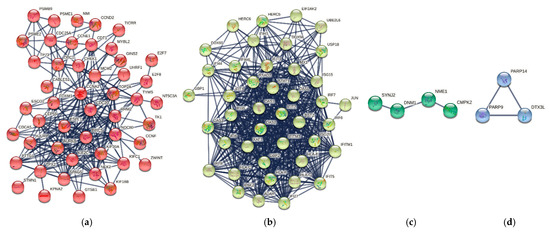
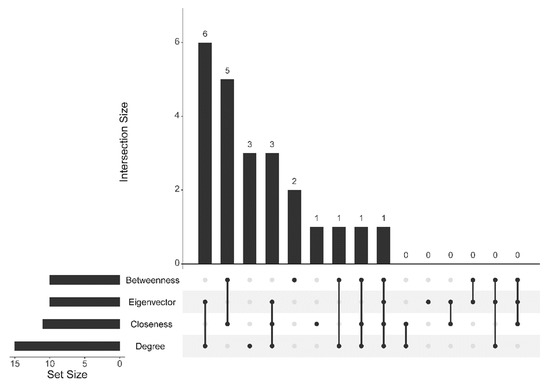
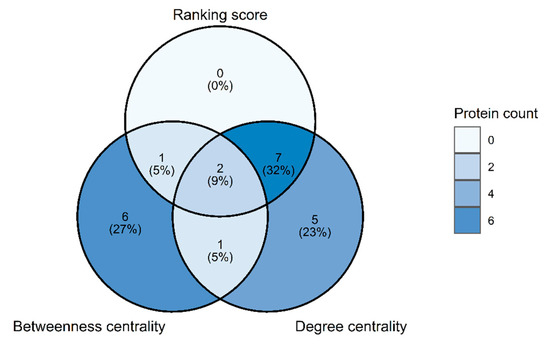
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}