Cystic Fibrosis: Cells, Physiopathology and Emerging Therapies
A topical collection in Cells (ISSN 2073-4409). This collection belongs to the section "Cellular Pathology".
Viewed by 87306
Share This Topical Collection
Editors
 Prof. Dr. Teresinha Leal
Prof. Dr. Teresinha Leal
Prof. Dr. Teresinha Leal
E-Mail
Website
Collection Editor
Louvain Centre for Toxicology and Applied Pharmacology, Institut de Recherche Expérimentale et Clinique, Université Catholique de Louvain, 1200 Brussels, Belgium
Interests: cystic fibrosis; cell physiology; ion transport; inflammatory responses; nasal potential difference; sweat test; mouse models; cell-based models
 Prof. Dr. Tristan Montier
Prof. Dr. Tristan Montier
Prof. Dr. Tristan Montier
E-Mail
Website
Collection Editor
1. Univ Brest, INSERM, EFS, UMR 1078, GGB, F-29200 Brest, France
2. CHRU de Brest, Service de Génétique Médicale et Biologie de la Reproduction, Centre de Référence des Maladies Rares “Maladies Neuromusculaires”, F-29200 Brest, France
Interests: gene transfer; human genomics; cystic fibrosis; nanoparticles; recombinant virus; lung delivery; aerosol; in vivo applications
Special Issues, Collections and Topics in MDPI journals
 Dr. Martial Delion
Dr. Martial Delion
Dr. Martial Delion
E-Mail
Website
Collection Editor
Laboratoire Rhéologie et Procédés, UMR5520. Université de Grenoble-Alpes, 38610 Gières, France
Interests: cystic fibrosis; cell biology; inflammation; airway mucus; mucins; polymorphonuclear neutrophils; CFTR modulation; rheology; optical tweezers; confocal and electronic microscopy
 Dr. Angélique Mottais
Dr. Angélique Mottais
Dr. Angélique Mottais
E-Mail
Website
Collection Editor
Louvain Centre for Toxicology and Applied Pharmacology, Institut de Recherche Expérimentale et Clinique, Université Catholique de Louvain, 1200 Brussels, Belgium
Interests: cystic fibrosis; gene therapy; cell physiology; inflammation; mouse models; cell-based models
Topical Collection Information
Dear Colleagues,
While lung disease remains the main cause of morbidity and mortality in cystic fibrosis (CF), it has been well established that this multisystemic genetic disease affects a large variety of organs and cell types. As a consequence, CF patients may develop several coexisting conditions, including diabetes, metabolic bone disorders, renal diseases, cancers, infertility or subfertility issues, and lung-transplantation-related complications. These comorbidities have become increasingly prevalent as CF patients live longer into adulthood. Even though CFTR has long been described as a chloride channel expressed at the apical membrane of epithelial cells, more recent studies have convincingly shown that in addition to airway epithelial cells, the protein is expressed in cells involved in inflammatory responses (neutrophils, macrophages, different subsets of lymphocytes, etc.), fibroblasts, pancreatic islet cells, bone cells, and many others. Therefore, it remains essential to improve our understanding of CF pathophysiology in each impacted cell type to develop new therapeutic strategies.
This present Topical Collection aims at shedding light on cell types and subtypes impacted by the presence of CFTR mutations; the value of cell-based models to study CF physiopathology and to quantify efficacy of emerging therapies; and how novel CF therapeutic strategies rescue CFTR-dependent cell processes.
We look forward to your contributions.
Prof. Dr. Teresinha Leal
Prof. Dr. Tristan Montier
Dr. Martial Delion
Dr. Angélique Mottais
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Cells is an international peer-reviewed open access semimonthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- cystic fibrosis
- CFTR
- cells
- pathophysiology
- inflammation
- interaction
- therapies
- cell-based models
Published Papers (18 papers)
Open AccessArticle
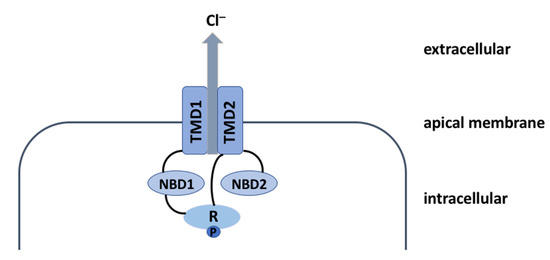
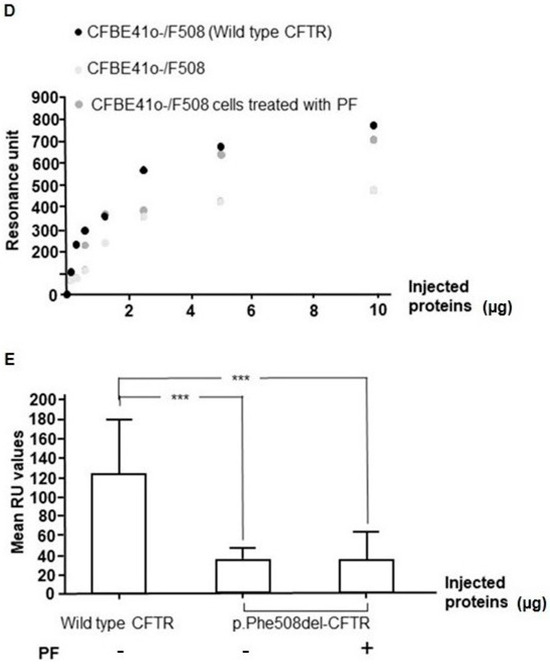
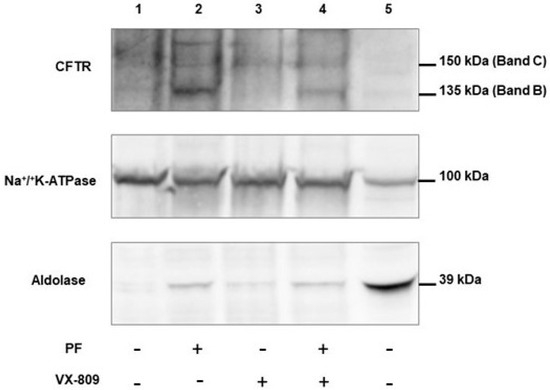
The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells
by
Raphaël Santinelli, Nathalie Benz, Julie Guellec, Fabien Quinquis, Ervin Kocas, Johan Thomas, Tristan Montier, Chandran Ka, Emilie Luczka-Majérus, Edouard Sage, Claude Férec, Christelle Coraux and Pascal Trouvé
Viewed by 1818
Abstract
Cystic Fibrosis (CF) is present due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, the most frequent variant being p.phe508del. The CFTR protein is a chloride (Cl-) channel which is defective and almost absent of cell membranes when the p.Phe508del
[...] Read more.
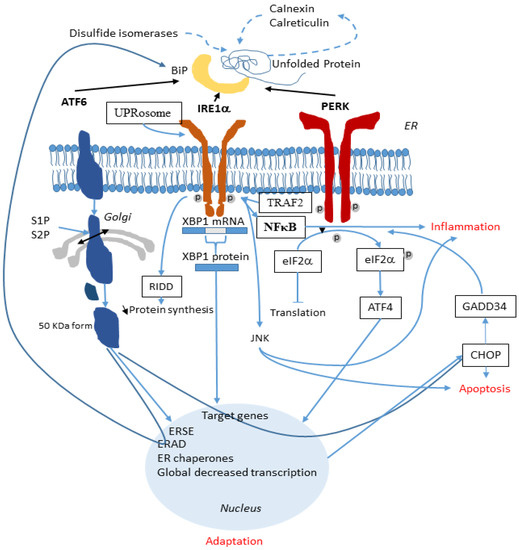
Cystic Fibrosis (CF) is present due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, the most frequent variant being p.phe508del. The CFTR protein is a chloride (Cl-) channel which is defective and almost absent of cell membranes when the p.Phe508del mutation is present. The p.Phe508del-CFTR protein is retained in the endoplasmic reticulum (ER) and together with inflammation and infection triggers the Unfolded Protein Response (UPR). During the UPR, the Activating Transcription Factor 6 (ATF6) is activated with cleavage and then decreases the expression of p.Phe508del-CFTR. We have previously shown that the inhibition of the activation of ATF6 alleviates the p.Phe508del-CFTR defects in cells overexpressing the mutated protein. In the present paper, our aim was to inhibit the cleavage of ATF6, and thus its activation in a human bronchial cell line with endogenous p.Phe508del-CFTR expression and in bronchial cells from patients, to be more relevant to CF. This was achieved by inhibiting the protease MBTP1 which is responsible for the cleavage of ATF6. We show here that this inhibition leads to increased mRNA and p.Phe508del-CFTR expression and, consequently, to increased Cl-efflux. We also explain the mechanisms linked to these increases with the modulation of genes when MBTP1 is inhibited. Indeed, RT-qPCR assays show that genes such as HSPA1B, CEBPB, VIMP, PFND2, MAPK8, XBP1, INSIG1, and CALR are modulated. In conclusion, we show that the inhibition of MBTP1 has a beneficial effect in relevant models to CF and that this is due to the modulation of genes involved in the disease.
Full article
►▼
Show Figures
Open AccessArticle
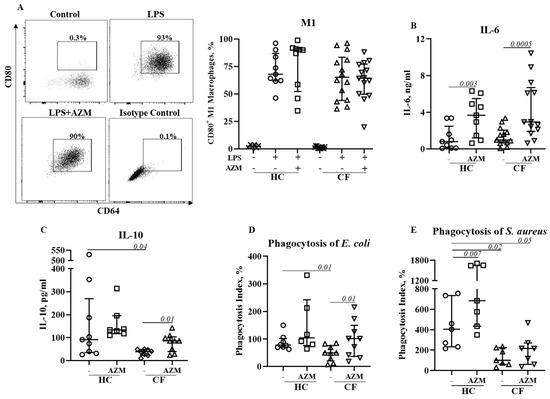
Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis
by
Abdullah A. Tarique, Neeraj Tuladhar, Dean Kelk, Nelufa Begum, Richard M. Lucas, Lin Luo, Jennifer L. Stow, Claire E. Wainwright, Scott C. Bell, Peter D. Sly and Emmanuelle Fantino
Viewed by 2086
Abstract
Background: Azithromycin (AZM) is widely being used for treating patients with cystic fibrosis (pwCF) following clinical trials demonstrating improved lung function and fewer incidents of pulmonary exacerba-tions. While the precise mechanisms remain elusive, immunomodulatory actions are thought to be involved. We previously reported
[...] Read more.
Background: Azithromycin (AZM) is widely being used for treating patients with cystic fibrosis (pwCF) following clinical trials demonstrating improved lung function and fewer incidents of pulmonary exacerba-tions. While the precise mechanisms remain elusive, immunomodulatory actions are thought to be involved. We previously reported impaired phagocytosis and defective anti-inflammatory M2 macrophage polarization in CF. This study systematically analyzed the effect of AZM on the functions of unpolarized and M1/M2 polarized macrophages in CF. Methods: Monocytes, isolated from the venous blood of patients with CF (pwCF) and healthy controls (HCs), were differentiated into monocyte-derived macrophages (MDMs) and subsequently infected with
P. aeruginosa.
P. aeruginosa uptake and killing by MDMs in the presence or absence of AZM was studied. M1 and M2 macrophage polarizations were induced and their functions and cytokine release were analyzed. Results: Following AZM treatment, both HC and CF MDMs exhibited a significant increase in
P. aeruginosa uptake and killing, however, lysosomal acidification remained unchanged. AZM treatment led to higher activation of ERK1/2 in both HC and CF MDMs. Pharmacological inhibition of ERK1/2 using U0126 significantly reduced
P. aeruginosa uptake in HC MDMs. M1 macrophage polarization remained unaffected; however, AZM treatment led to increased IL-6 and IL-10 release in both HC and CF M1 macrophages. AZM also significantly increased the phagocytic index for both pHrodo
E. coli and
S. aureus in CF M1 macrophages. In CF, AZM treatment promoted anti-inflammatory M2 macrophage polarization, with an increased percentage of CD209
+ M2 macrophages, induction of the M2 gene
CCL18, along with its secretion in the culture supernatant. However, AZM d’d not restore endocytosis in CF, another essential feature of M2 macrophages. Conclusions: This study highlights the cellular functions and molecular targets of AZM which may involve an improved uptake of both Gram-positive and Gram-negative bacteria, restored anti-inflammatory macrophage polarization in CF. This may in turn shape the reduced lung inflammation observed in clinical trials. In addition, we confirmed the role of ERK1/2 activation for bacterial uptake.
Full article
►▼
Show Figures
Open AccessArticle
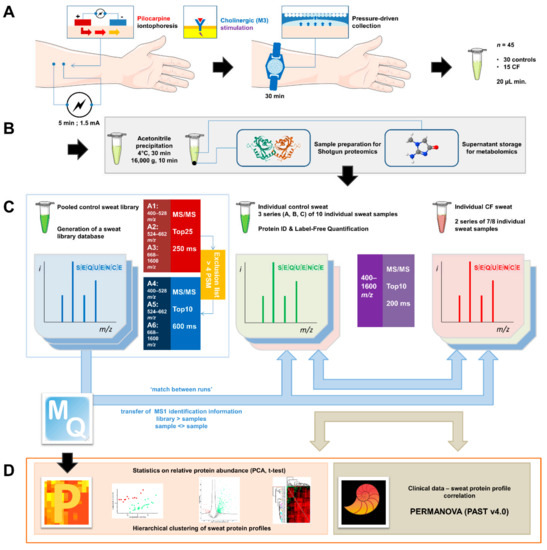
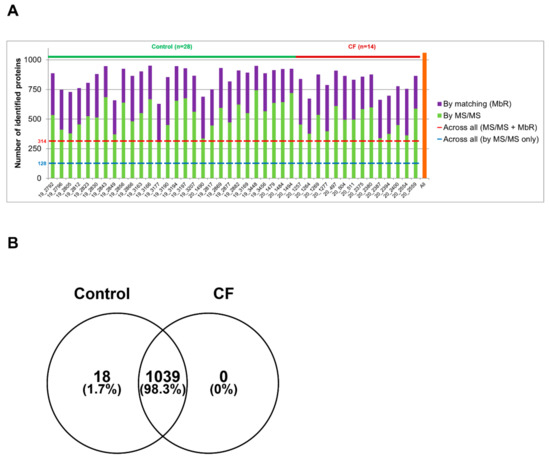
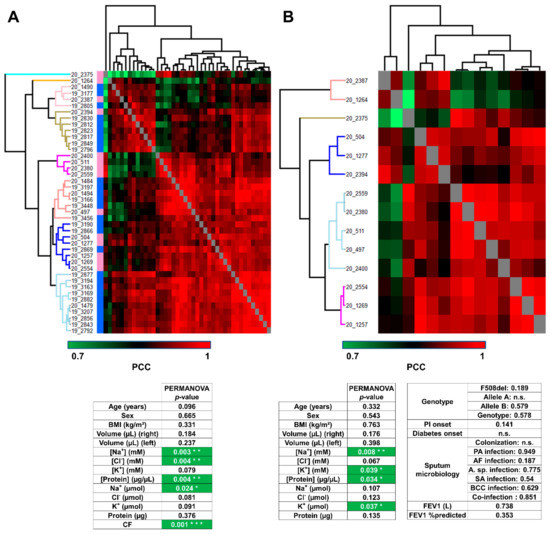
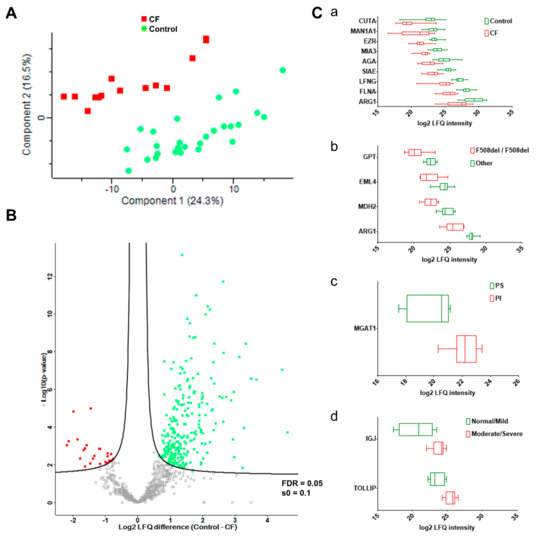
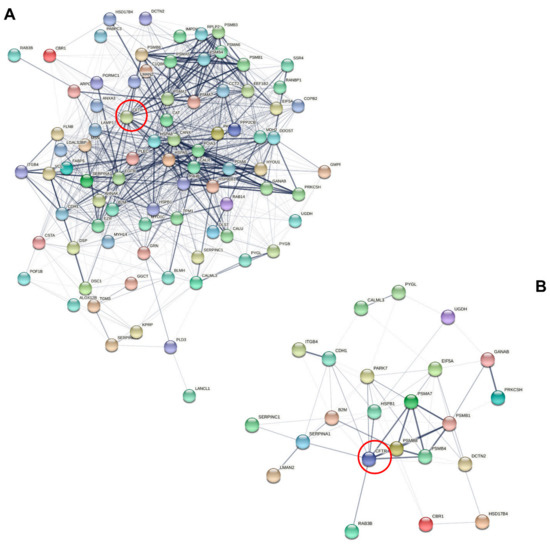
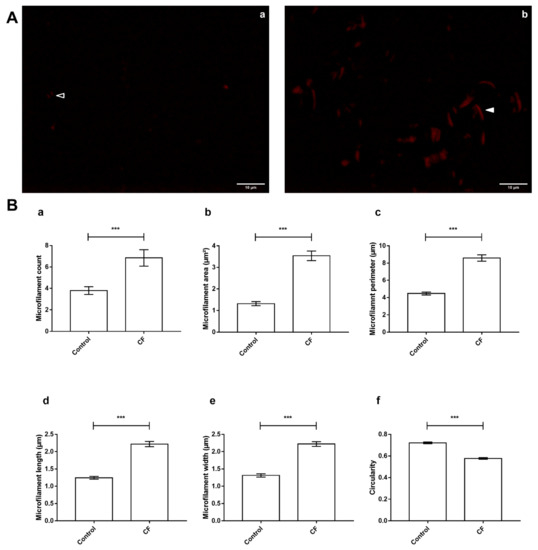
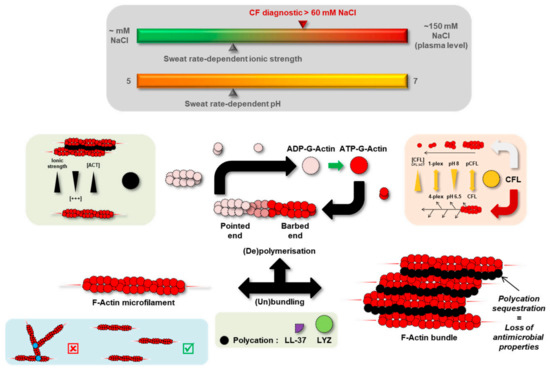
Sweat Proteomics in Cystic Fibrosis: Discovering Companion Biomarkers for Precision Medicine and Therapeutic Development
by
Bastien Burat, Audrey Reynaerts, Dominique Baiwir, Maximilien Fléron, Sophie Gohy, Gauthier Eppe, Teresinha Leal and Gabriel Mazzucchelli
Cited by 4 | Viewed by 3203
Abstract
In clinical routine, the diagnosis of cystic fibrosis (CF) is still challenging regardless of international consensus on diagnosis guidelines and tests. For decades, the classical Gibson and Cooke test measuring sweat chloride concentration has been a keystone, yet, it may provide normal or
[...] Read more.
In clinical routine, the diagnosis of cystic fibrosis (CF) is still challenging regardless of international consensus on diagnosis guidelines and tests. For decades, the classical Gibson and Cooke test measuring sweat chloride concentration has been a keystone, yet, it may provide normal or equivocal results. As of now, despite the combination of sweat testing,
CFTR genotyping, and CFTR functional testing, a small fraction (1–2%) of inconclusive diagnoses are reported and justifies the search for new CF biomarkers. More importantly, in the context of precision medicine, with a view to early diagnosis, better prognosis, appropriate clinical follow-up, and new therapeutic development, discovering companion biomarkers of CF severity and phenotypic rescue are of utmost interest. To date, previous sweat proteomic studies have already documented disease-specific variations of sweat proteins (e.g., in schizophrenia and tuberculosis). In the current study, sweat samples from 28 healthy control subjects and 14 patients with CF were analyzed by nanoUHPLC-Q-Orbitrap-based shotgun proteomics, to look for CF-associated changes in sweat protein composition and abundance. A total of 1057 proteins were identified and quantified at an individual level, by a shotgun label-free approach. Notwithstanding similar proteome composition, enrichment, and functional annotations, control and CF samples featured distinct quantitative proteome profiles significantly correlated with CF, accounting for the respective inter-individual variabilities of control and CF sweat. All in all: (i) 402 sweat proteins were differentially abundant between controls and patients with CF, (ii) 68 proteins varied in abundance between F508del homozygous patients and patients with another genotype, (iii) 71 proteins were differentially abundant according to the pancreatic function, and iv) 54 proteins changed in abundance depending on the lung function. The functional annotation of pathophysiological biomarkers highlighted eccrine gland cell perturbations in: (i) protein biosynthesis and trafficking, (ii) CFTR proteostasis and membrane stability, and (iii) cell-cell adherence, membrane integrity, and cytoskeleton crosstalk. Cytoskeleton-related biomarkers were of utmost interest because of the consistency between variations observed here in CF sweat and variations previously documented in other CF tissues. From a clinical stance, nine candidate biomarkers of CF diagnosis (CUTA, ARG1, EZR, AGA, FLNA, MAN1A1, MIA3, LFNG, SIAE) and seven candidate biomarkers of CF severity (ARG1, GPT, MDH2, EML4 (F508del homozygous), MGAT1 (pancreatic insufficiency), IGJ, TOLLIP (lung function impairment)) were deemed suitable for further verification.
Full article
►▼
Show Figures
Open AccessReview
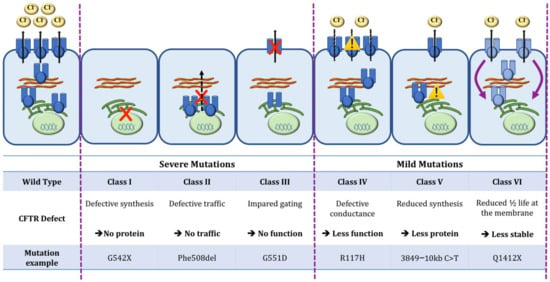
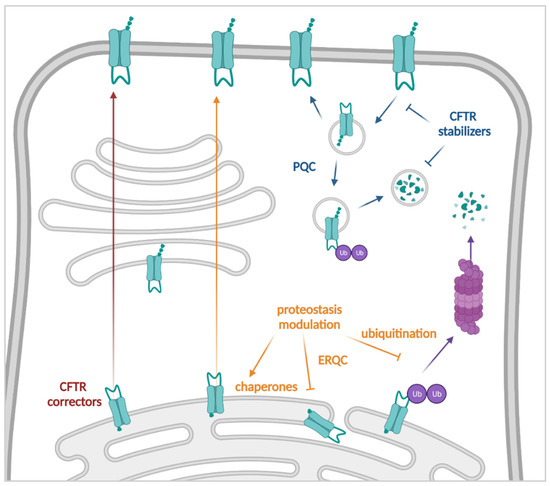
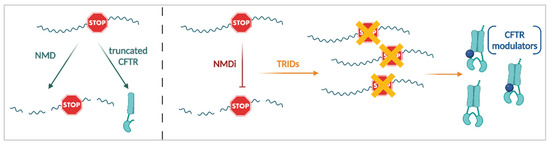
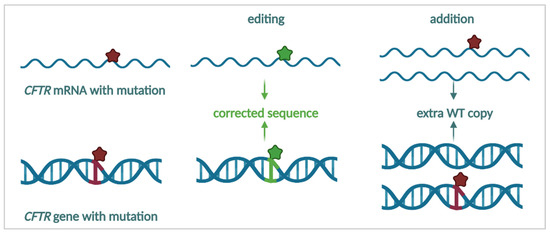
One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies
by
Marjolein M. Ensinck and Marianne S. Carlon
Cited by 19 | Viewed by 5982
Abstract
Cystic fibrosis (CF) is the most common monogenic disorder, caused by mutations in the CF transmembrane conductance regulator (
CFTR) gene. Over the last 30 years, tremendous progress has been made in understanding the molecular basis of CF and the development of
[...] Read more.
Cystic fibrosis (CF) is the most common monogenic disorder, caused by mutations in the CF transmembrane conductance regulator (
CFTR) gene. Over the last 30 years, tremendous progress has been made in understanding the molecular basis of CF and the development of treatments that target the underlying defects in CF. Currently, a highly effective CFTR modulator treatment (Kalydeco™/Trikafta™) is available for 90% of people with CF. In this review, we will give an extensive overview of past and ongoing efforts in the development of therapies targeting the molecular defects in CF. We will discuss strategies targeting the CFTR protein (i.e., CFTR modulators such as correctors and potentiators), its cellular environment (i.e., proteostasis modulation, stabilization at the plasma membrane), the
CFTR mRNA (i.e., amplifiers, nonsense mediated mRNA decay suppressors, translational readthrough inducing drugs) or the
CFTR gene (gene therapies). Finally, we will focus on how these efforts can be applied to the 15% of people with CF for whom no causal therapy is available yet.
Full article
►▼
Show Figures
Open AccessReview
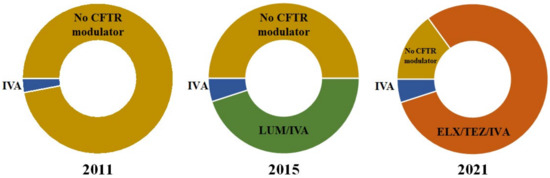
CFTR Modulators in People with Cystic Fibrosis: Real-World Evidence in France
by
Lucile Regard, Clémence Martin, Espérie Burnet, Jennifer Da Silva and Pierre-Régis Burgel
Cited by 30 | Viewed by 6742
Abstract
Cystic fibrosis (CF) is a rare genetic multisystemic disease, the manifestations of which are due to mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein and can lead to respiratory insufficiency and premature death. CFTR modulators, which were developed in
[...] Read more.
Cystic fibrosis (CF) is a rare genetic multisystemic disease, the manifestations of which are due to mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein and can lead to respiratory insufficiency and premature death. CFTR modulators, which were developed in the past decade, partially restore CFTR protein function. Their clinical efficacy has been demonstrated in phase 3 clinical trials, particularly in terms of lung function and pulmonary exacerbations, nutritional status, and quality of life in people with gating mutations (ivacaftor), homozygous for the F508del mutation (lumacaftor/ivacaftor and tezacaftor/ivacaftor), and in those with at least one F508del mutation (elexacaftor/tezacaftor/ivacaftor). However, many questions remain regarding their long-term safety and effectiveness, particularly in patients with advanced lung disease, liver disease, renal insufficiency, or problematic bacterial colonization. The impact of CFTR modulators on other important outcomes such as concurrent treatments, lung transplantation, chest imaging, or pregnancies also warrants further investigation. The French CF Reference Network includes 47 CF centers that contribute patient data to the comprehensive French CF Registry and have conducted nationwide real-world studies on CFTR modulators. This review seeks to summarize the results of these real-world studies and examine their findings against those of randomized control trials.
Full article
►▼
Show Figures
Open AccessArticle
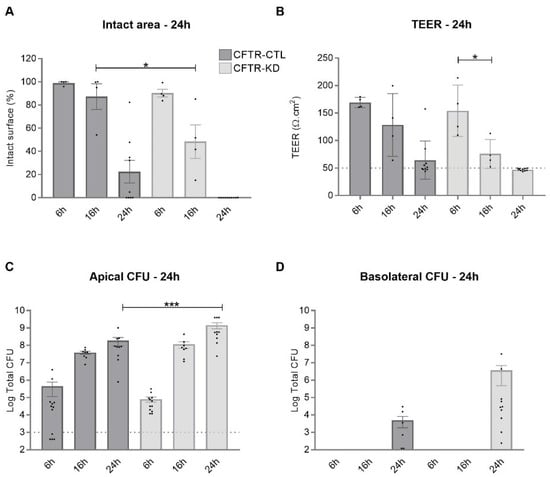
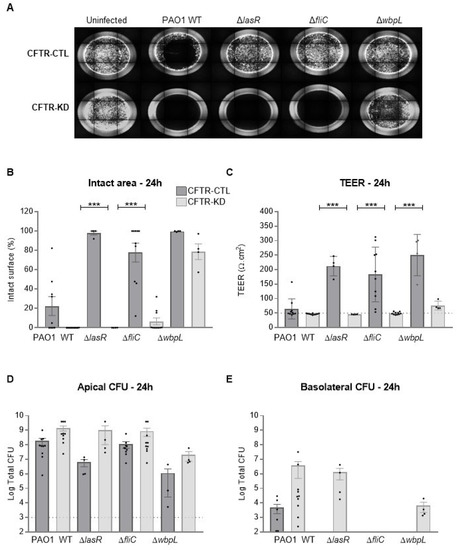
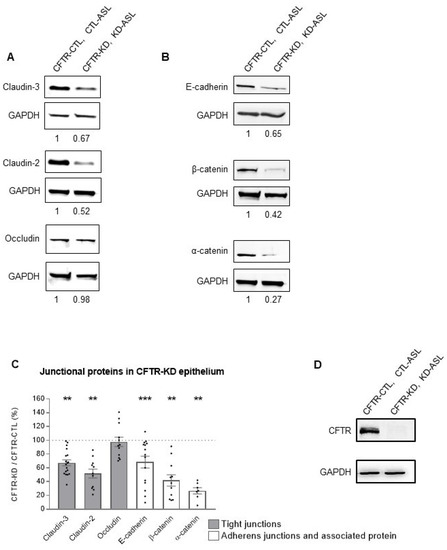
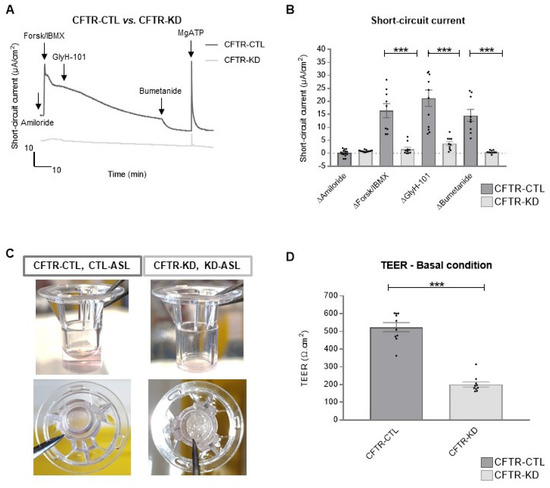
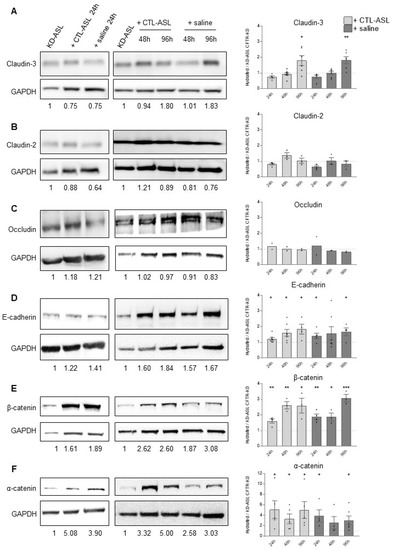
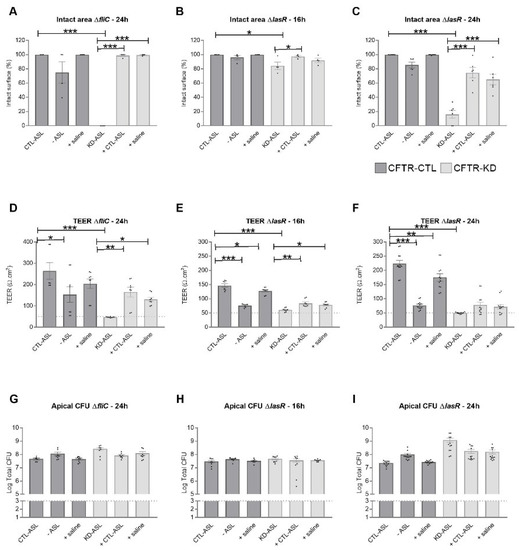
Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks
by
Juliette L. Simonin, Alexandre Luscher, Davide Losa, Mehdi Badaoui, Christian van Delden, Thilo Köhler and Marc Chanson
Cited by 8 | Viewed by 6078
Abstract
Defective hydration of airway surface mucosa is associated with recurrent lung infection in cystic fibrosis (CF), a disease caused by CF transmembrane conductance regulator (
CFTR) gene mutations. Whether the composition and/or presence of an airway surface liquid (ASL) is sufficient to
[...] Read more.
Defective hydration of airway surface mucosa is associated with recurrent lung infection in cystic fibrosis (CF), a disease caused by CF transmembrane conductance regulator (
CFTR) gene mutations. Whether the composition and/or presence of an airway surface liquid (ASL) is sufficient to prevent infection remains unclear. The susceptibility to infection of polarized wild type and
CFTR knockdown (CFTR-KD) airway epithelial cells was determined in the presence or absence of a healthy ASL or physiological saline. CFTR-KD epithelia exhibited strong ASL volume reduction, enhanced susceptibility to infection, and reduced junctional integrity. Interestingly, the presence of an apical physiological saline alleviated disruption of the airway epithelial barrier by stimulating essential junctional protein expression. Thus, rehydrated CFTR-KD cells were protected from infection despite normally intense bacterial growth. This study indicates that an epithelial integrity gatekeeper is modulated by the presence of an apical liquid volume, irrespective of the liquid’s composition and of expression of a functional CFTR.
Full article
►▼
Show Figures
Open AccessReview
CFTR Modulator Therapies: Potential Impact on Airway Infections in Cystic Fibrosis
by
Francesca Saluzzo, Luca Riberi, Barbara Messore, Nicola Ivan Loré, Irene Esposito, Elisabetta Bignamini and Virginia De Rose
Cited by 22 | Viewed by 4085
Abstract
Cystic Fibrosis (CF) is an autosomal recessive disease caused by mutations in the gene encoding for the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) protein, expressed on the apical surface of epithelial cells. CFTR absence/dysfunction results in ion imbalance and airway surface dehydration that
[...] Read more.
Cystic Fibrosis (CF) is an autosomal recessive disease caused by mutations in the gene encoding for the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) protein, expressed on the apical surface of epithelial cells. CFTR absence/dysfunction results in ion imbalance and airway surface dehydration that severely compromise the CF airway microenvironment, increasing infection susceptibility. Recently, novel therapies aimed at correcting the basic CFTR defect have become available, leading to substantial clinical improvement of CF patients. The restoration or increase of CFTR function affects the airway microenvironment, improving local defence mechanisms. CFTR modulator drugs might therefore affect the development of chronic airway infections and/or improve the status of existing infections in CF. Thus far, however, the full extent of these effects of CFTR-modulators, especially in the long-term remains still unknown. This review aims to provide an overview of current evidence on the potential impact of CFTR modulators on airway infections in CF. Their role in affecting CF microbiology, the susceptibility to infections as well as the potential efficacy of their use in preventing/decreasing the development of chronic lung infections and the recurrent acute exacerbations in CF will be critically analysed.
Full article
Open AccessArticle
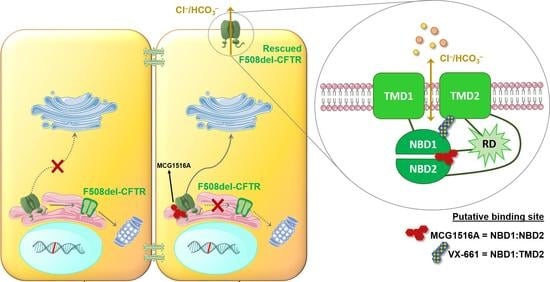
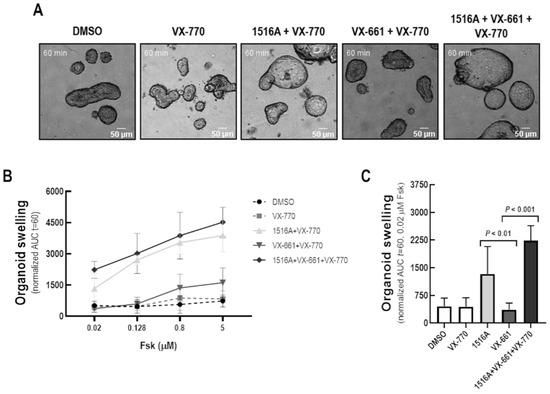
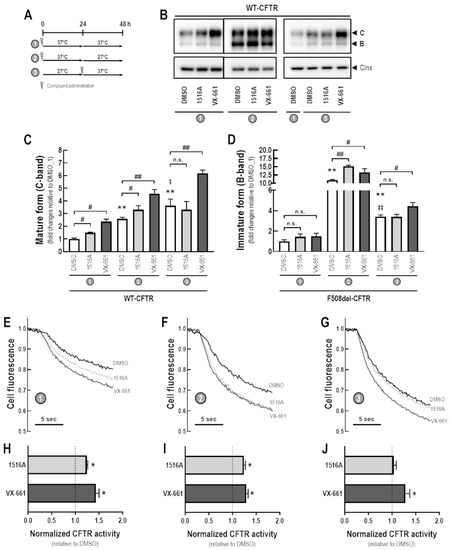
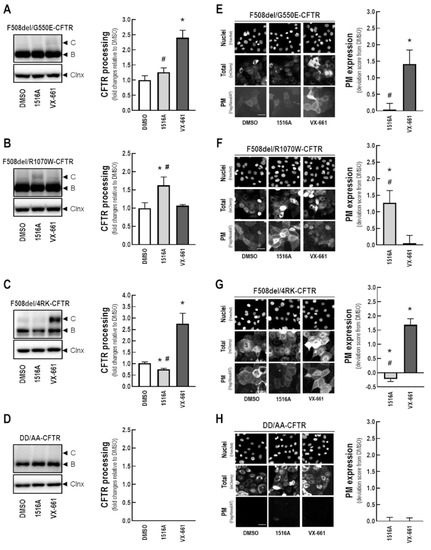
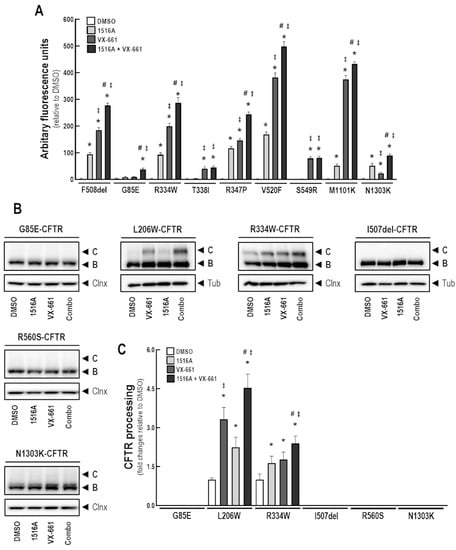
Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A
by
Miquéias Lopes-Pacheco, Mafalda Bacalhau, Sofia S. Ramalho, Iris A. L. Silva, Filipa C. Ferreira, Graeme W. Carlile, David Y. Thomas, Carlos M. Farinha, John W. Hanrahan and Margarida D. Amaral
Cited by 11 | Viewed by 5411
Abstract
Although some therapeutic progress has been achieved in developing small molecules that correct F508del-CFTR defects, the mechanism of action (MoA) of these compounds remain poorly elucidated. Here, we investigated the effects and MoA of MCG1516A, a newly developed F508del-CFTR corrector. MCG1516A effects on
[...] Read more.
Although some therapeutic progress has been achieved in developing small molecules that correct F508del-CFTR defects, the mechanism of action (MoA) of these compounds remain poorly elucidated. Here, we investigated the effects and MoA of MCG1516A, a newly developed F508del-CFTR corrector. MCG1516A effects on wild-type (WT) and F508del-CFTR were assessed by immunofluorescence microscopy, and biochemical and functional assays both in cell lines and in intestinal organoids. To shed light on the MoA of MCG1516A, we evaluated its additivity to the FDA-approved corrector VX-661, low temperature, genetic revertants of F508del-CFTR (G550E, R1070W, and 4RK), and the traffic-null variant DD/AA. Finally, we explored the ability of MCG1516A to rescue trafficking and function of other CF-causing mutations. We found that MCG1516A rescues F508del-CFTR with additive effects to VX-661. A similar behavior was observed for WT-CFTR. Under low temperature incubation, F508del-CFTR demonstrated an additivity in processing and function with VX-661, but not with MCG1516A. In contrast, both compounds promoted additional effects to low temperature to WT-CFTR. MCG1516A demonstrated additivity to genetic revertant R1070W, while VX-661 was additive to G550E and 4RK. Nevertheless, none of these compounds rescued DD/AA trafficking. Both MCG1516A and VX-661 rescued CFTR processing of L206W- and R334W-CFTR with greater effects when these compounds were combined. In summary, the absence of additivity of MCG1516A to genetic revertant G550E suggests a putative binding site for this compound on NBD1:NBD2 interface. Therefore, a combination of MCG1516A with compounds able to rescue DD/AA traffic, or mimicking the actions of revertant R1070W (e.g., VX-661), could enhance correction of F508del-CFTR defects.
Full article
►▼
Show Figures
Open AccessReview
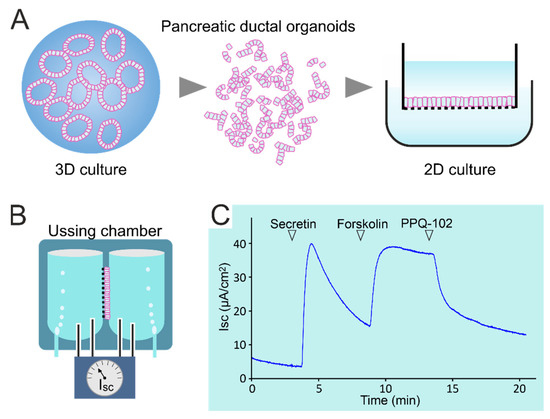
Bicarbonate Transport in Cystic Fibrosis and Pancreatitis
by
Dora Angyal, Marcel J. C. Bijvelds, Marco J. Bruno, Maikel P. Peppelenbosch and Hugo R. de Jonge
Cited by 19 | Viewed by 8294
Abstract
CFTR, the cystic fibrosis (CF) gene-encoded epithelial anion channel, has a prominent role in driving chloride, bicarbonate and fluid secretion in the ductal cells of the exocrine pancreas. Whereas severe mutations in
CFTR cause fibrosis of the pancreas in utero, CFTR mutants with
[...] Read more.
CFTR, the cystic fibrosis (CF) gene-encoded epithelial anion channel, has a prominent role in driving chloride, bicarbonate and fluid secretion in the ductal cells of the exocrine pancreas. Whereas severe mutations in
CFTR cause fibrosis of the pancreas in utero, CFTR mutants with residual function, or CFTR variants with a normal chloride but defective bicarbonate permeability (CFTR
BD), are associated with an enhanced risk of pancreatitis. Recent studies indicate that CFTR function is not only compromised in genetic but also in selected patients with an acquired form of pancreatitis induced by alcohol, bile salts or smoking. In this review, we summarize recent insights into the mechanism and regulation of CFTR-mediated and modulated bicarbonate secretion in the pancreatic duct, including the role of the osmotic stress/chloride sensor WNK1 and the scaffolding protein IRBIT, and current knowledge about the role of CFTR in genetic and acquired forms of pancreatitis. Furthermore, we discuss the perspectives for CFTR modulator therapy in the treatment of exocrine pancreatic insufficiency and pancreatitis and introduce pancreatic organoids as a promising model system to study CFTR function in the human pancreas, its role in the pathology of pancreatitis and its sensitivity to CFTR modulators on a personalized basis.
Full article
►▼
Show Figures
Open AccessReview
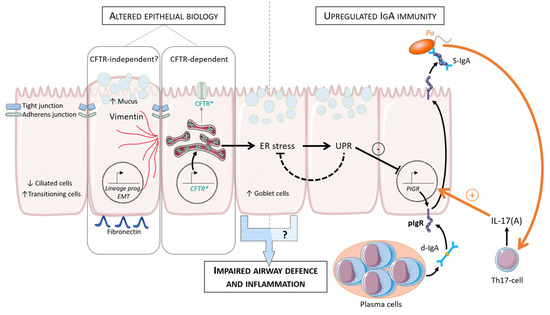
Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis
by
Sophie Gohy, Alexandra Moeremans, Charles Pilette and Amandine Collin
Cited by 10 | Viewed by 4258
Abstract
The respiratory epithelium represents the first chemical, immune, and physical barrier against inhaled noxious materials, particularly pathogens in cystic fibrosis. Local mucus thickening, altered mucociliary clearance, and reduced pH due to CFTR protein dysfunction favor bacterial overgrowth and excessive inflammation. We aimed in
[...] Read more.
The respiratory epithelium represents the first chemical, immune, and physical barrier against inhaled noxious materials, particularly pathogens in cystic fibrosis. Local mucus thickening, altered mucociliary clearance, and reduced pH due to CFTR protein dysfunction favor bacterial overgrowth and excessive inflammation. We aimed in this review to summarize respiratory mucosal alterations within the epithelium and current knowledge on local immunity linked to immunoglobulin A in patients with cystic fibrosis.
Full article
►▼
Show Figures
Open AccessReview
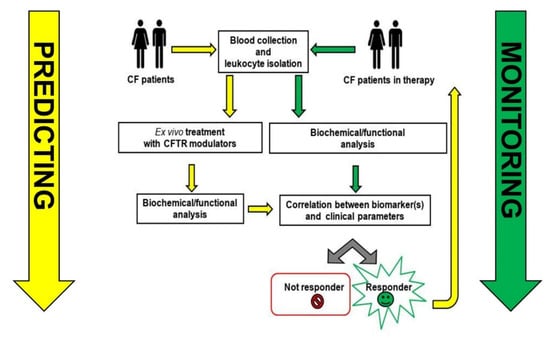
Revisiting the Role of Leukocytes in Cystic Fibrosis
by
Monica Averna, Paola Melotti and Claudio Sorio
Cited by 12 | Viewed by 3141
Abstract
Cystic fibrosis in characterized by pulmonary bacterial colonization and hyperinflammation. Lymphocytes, monocytes/macrophages, neutrophils, and dendritic cells of patients with CF express functional CFTR and are directly affected by altered CFTR expression/function, impairing their ability to resolve infections and inflammation. However, the mechanism behind
[...] Read more.
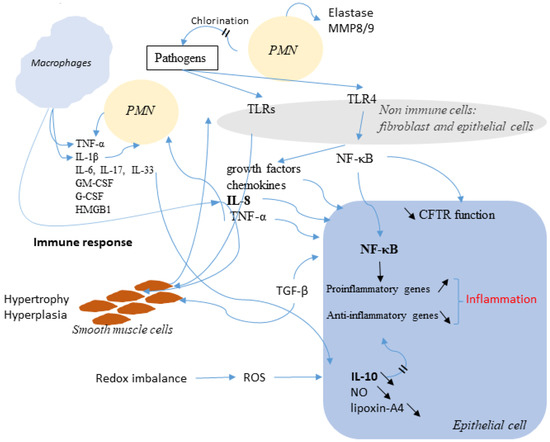
Cystic fibrosis in characterized by pulmonary bacterial colonization and hyperinflammation. Lymphocytes, monocytes/macrophages, neutrophils, and dendritic cells of patients with CF express functional CFTR and are directly affected by altered CFTR expression/function, impairing their ability to resolve infections and inflammation. However, the mechanism behind and the contribution of leukocytes in the pathogenesis of CF are still poorly characterized. The recent clinical introduction of specific CFTR modulators added an important tool not only for the clinical management of the disease but also to the investigation of the pathophysiological mechanisms related to CFTR dysfunction and dysregulated immunity. These drugs treat the basic defect in cystic fibrosis (CF) by increasing CFTR function with improvement of lung function and quality of life, and may improve clinical outcomes also by correcting the dysregulated immune function that characterizes CF. Measure of CFTR function, protein expression profiling and several omics methods were used to identify molecular changes in freshly isolated leukocytes of CF patients, highlighting two roles of leukocytes in CF: one more generally related to the mechanism(s) causing immune dysregulation in CF and unresolved inflammation, and another more applicative role, which identifies in myeloid cells, an important tool predictive of the therapeutic response of CF patients. In this review we will summarize available data on CFTR expression and function in leukocyte populations and will discuss potential clinical applications based on available data.
Full article
►▼
Show Figures
Open AccessReview
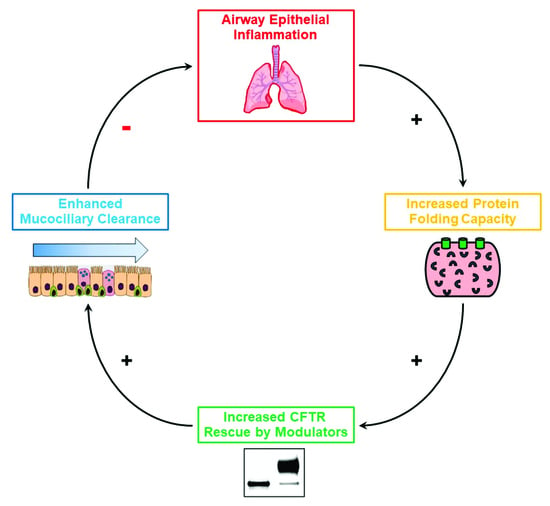
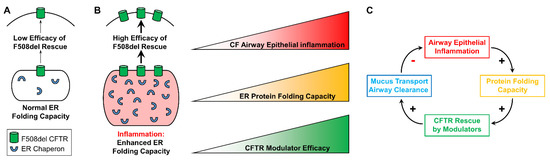
Impact of Airway Inflammation on the Efficacy of CFTR Modulators
by
Carla M. P. Ribeiro and Martina Gentzsch
Cited by 14 | Viewed by 3004
Abstract
Defective CFTR biogenesis and activity in cystic fibrosis airways leads to airway dehydration and impaired mucociliary clearance, resulting in chronic airway infection and inflammation. Most cystic fibrosis patients have at least one copy of the F508del CFTR mutation, which results in a protein
[...] Read more.
Defective CFTR biogenesis and activity in cystic fibrosis airways leads to airway dehydration and impaired mucociliary clearance, resulting in chronic airway infection and inflammation. Most cystic fibrosis patients have at least one copy of the F508del CFTR mutation, which results in a protein retained in the endoplasmic reticulum and degraded by the proteosomal pathway. CFTR modulators, e.g., correctors, promote the transfer of F508del to the apical membrane, while potentiators increase CFTR activity. Corrector and potentiator double therapies modestly improve lung function, whereas triple therapies with two correctors and one potentiator indicate improved outcomes. Enhanced F508del rescue by CFTR modulators is achieved by exposing F508del/F508del primary cultures of human bronchial epithelia to relevant inflammatory stimuli, i.e., supernatant from mucopurulent material or bronchoalveolar lavage fluid from human cystic fibrosis airways. Inflammation enhances the biochemical and functional rescue of F508del by double or triple CFTR modulator therapy and overcomes abrogation of CFTR correction by chronic VX-770 treatment in vitro. Furthermore, the impact of inflammation on clinical outcomes linked to CFTR rescue has been recently suggested. This review discusses these data and possible mechanisms for airway inflammation-enhanced F508del rescue. Expanding the understanding of how airway inflammation improves CFTR rescue may benefit cystic fibrosis patients.
Full article
►▼
Show Figures
Open AccessArticle
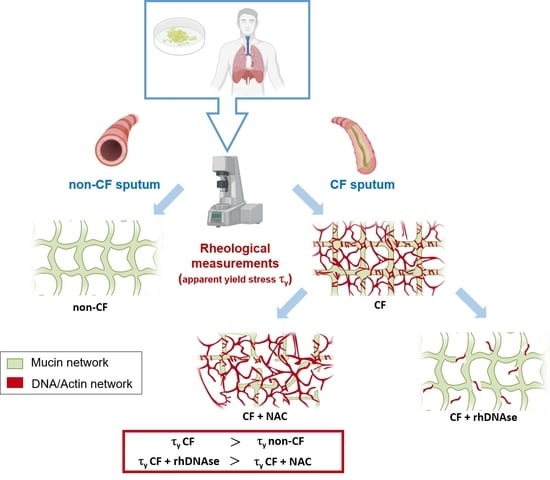
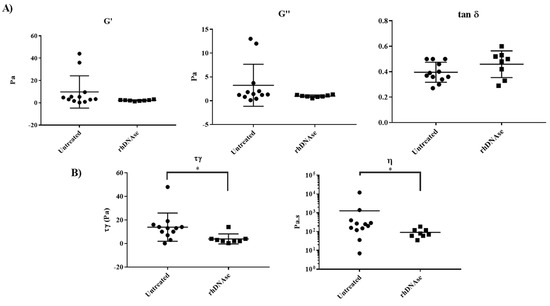
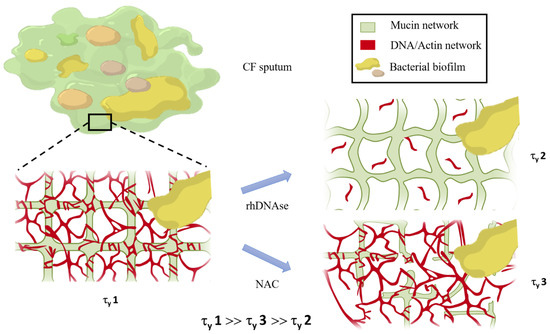
Apparent Yield Stress of Sputum as a Relevant Biomarker in Cystic Fibrosis
by
Rosy Ghanem, Philippe Roquefort, Sophie Ramel, Véronique Laurent, Tanguy Haute, Tony Le Gall, Thierry Aubry and Tristan Montier
Cited by 10 | Viewed by 2596
Abstract
The mucus obstructing the airways of Cystic Fibrosis (CF) patients is a yield stress fluid. Linear and non-linear rheological analyses of CF sputa can provide relevant biophysical markers, which could be used for the management of this disease. Sputa were collected from CF
[...] Read more.
The mucus obstructing the airways of Cystic Fibrosis (CF) patients is a yield stress fluid. Linear and non-linear rheological analyses of CF sputa can provide relevant biophysical markers, which could be used for the management of this disease. Sputa were collected from CF patients either without any induction or following an aerosol treatment with the recombinant human DNAse (rhDNAse, Pulmozyme
®). Several sample preparations were considered and multiple measurements were performed in order to assess both the repeatability and the robustness of the rheological measurements. The linear and non-linear rheological properties of all CF sputa were characterized. While no correlation between oscillatory shear linear viscoelastic properties and clinical data was observed, the steady shear flow data showed that the apparent yield stress of sputum from CF patients previously treated with rhDNAse was approximately one decade lower than that of non-treated CF patients. Similar results were obtained with sputa from non-induced CF patients subjected ex vivo to a Pulmozyme
® aerosol treatment. The results demonstrate that the apparent yield stress of patient sputa is a relevant predictive/prognostic biomarker in CF patients and could help in the development of new mucolytic agents.
Full article
►▼
Show Figures
Open AccessReview
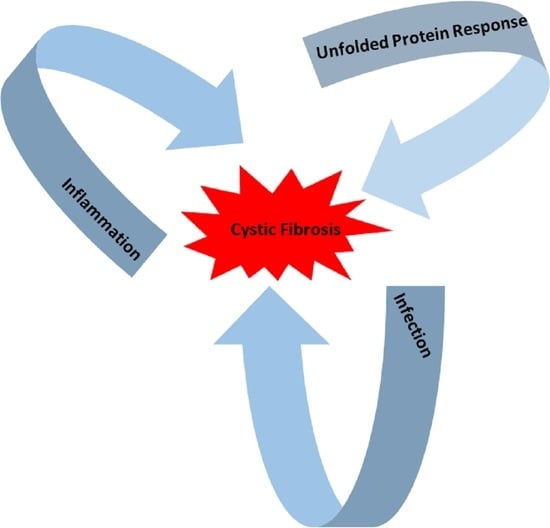
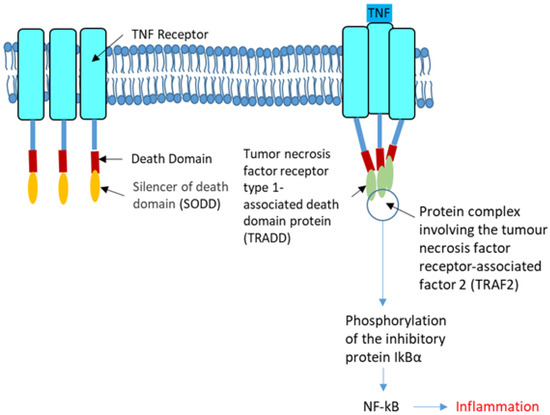
The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis
by
Pascal Trouvé, Claude Férec and Emmanuelle Génin
Cited by 15 | Viewed by 3311
Abstract
In cystic fibrosis (CF), p.Phe508del is the most frequent mutation in the Cystic Fibrosis Transmembrane conductance Regulator (
CFTR) gene. The p.Phe508del-CFTR protein is retained in the ER and rapidly degraded. This retention likely triggers an atypical Unfolded Protein Response (UPR) involving
[...] Read more.
In cystic fibrosis (CF), p.Phe508del is the most frequent mutation in the Cystic Fibrosis Transmembrane conductance Regulator (
CFTR) gene. The p.Phe508del-CFTR protein is retained in the ER and rapidly degraded. This retention likely triggers an atypical Unfolded Protein Response (UPR) involving ATF6, which reduces the expression of p.Phe508del-CFTR. There are still some debates on the role of the UPR in CF: could it be triggered by the accumulation of misfolded CFTR proteins in the endoplasmic reticulum as was proposed for the most common CFTR mutation p.Phe508del? Or, is it the consequence of inflammation and infection that occur in the disease? In this review, we summarize recent findings on UPR in CF and show how infection, inflammation and UPR act together in CF. We propose to rethink their respective role in CF and to consider them as a whole.
Full article
►▼
Show Figures
Open AccessReview
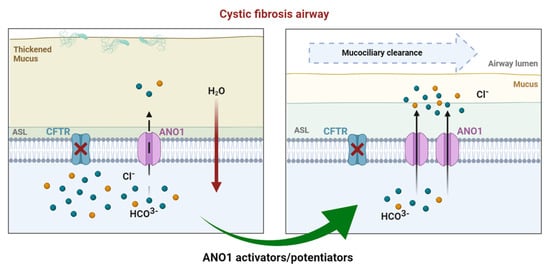
TMEM16A/ANO1: Current Strategies and Novel Drug Approaches for Cystic Fibrosis
by
Christie Mitri, Himanshu Sharma, Harriet Corvol and Olivier Tabary
Cited by 9 | Viewed by 4358
Abstract
Cystic fibrosis (CF) is the most common of rare hereditary diseases in Caucasians, and it is estimated to affect 75,000 patients globally. CF is a complex disease due to the multiplicity of mutations found in the CF transmembrane conductance regulator (CFTR) gene causing
[...] Read more.
Cystic fibrosis (CF) is the most common of rare hereditary diseases in Caucasians, and it is estimated to affect 75,000 patients globally. CF is a complex disease due to the multiplicity of mutations found in the CF transmembrane conductance regulator (CFTR) gene causing the CFTR protein to become dysfunctional. Correctors and potentiators have demonstrated good clinical outcomes for patients with specific gene mutations; however, there are still patients for whom those treatments are not suitable and require alternative CFTR-independent strategies. Although CFTR is the main chloride channel in the lungs, others could, e.g., anoctamin-1 (ANO1 or TMEM16A), compensate for the deficiency of CFTR. This review summarizes the current knowledge on calcium-activated chloride channel (CaCC) ANO1 and presents ANO1 as an exciting target in CF.
Full article
►▼
Show Figures
Open AccessFeature PaperReview
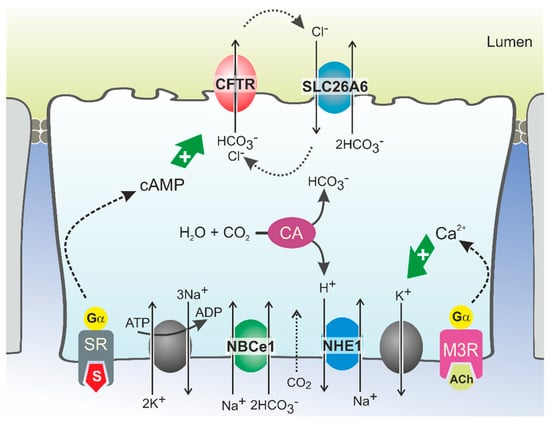
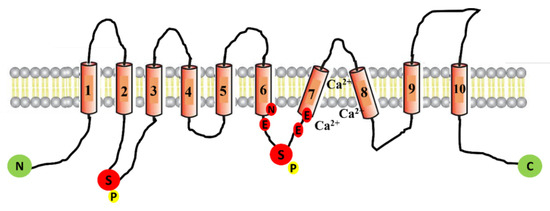
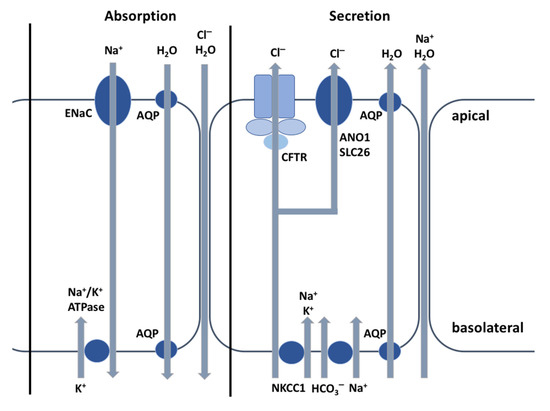
CFTR Protein: Not Just a Chloride Channel?
by
Laurence S. Hanssens, Jean Duchateau and Georges J. Casimir
Cited by 57 | Viewed by 11596
Abstract
Cystic fibrosis (CF) is a recessive genetic disease caused by mutations in a gene encoding a protein called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). The CFTR protein is known to acts as a chloride (Cl
−) channel expressed in the exocrine glands
[...] Read more.
Cystic fibrosis (CF) is a recessive genetic disease caused by mutations in a gene encoding a protein called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). The CFTR protein is known to acts as a chloride (Cl
−) channel expressed in the exocrine glands of several body systems where it also regulates other ion channels, including the epithelial sodium (Na
+) channel (ENaC) that plays a key role in salt absorption. This function is crucial to the osmotic balance of the mucus and its viscosity. However, the pathophysiology of CF is more challenging than a mere dysregulation of epithelial ion transport, mainly resulting in impaired mucociliary clearance (MCC) with consecutive bronchiectasis and in exocrine pancreatic insufficiency. This review shows that the CFTR protein is not just a chloride channel. For a long time, research in CF has focused on abnormal Cl
− and Na
+ transport. Yet, the CFTR protein also regulates numerous other pathways, such as the transport of HCO
3−, glutathione and thiocyanate, immune cells, and the metabolism of lipids. It influences the pH homeostasis of airway surface liquid and thus the MCC as well as innate immunity leading to chronic infection and inflammation, all of which are considered as key pathophysiological characteristics of CF.
Full article
►▼
Show Figures
Open AccessReview
Therapeutic Approaches for Patients with Cystic Fibrosis Not Eligible for Current CFTR Modulators
by
Isabelle Fajac and Isabelle Sermet
Cited by 25 | Viewed by 4994
Abstract
Cystic fibrosis is a severe autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (
CFTR) gene encoding the CFTR protein, a chloride channel expressed in many epithelial cells. New drugs called CFTR modulators aim at restoring the
[...] Read more.
Cystic fibrosis is a severe autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (
CFTR) gene encoding the CFTR protein, a chloride channel expressed in many epithelial cells. New drugs called CFTR modulators aim at restoring the CFTR protein function, and they will benefit many patients with cystic fibrosis in the near future. However, some patients bear rare mutations that are not yet eligible for CFTR modulators, although they might be amenable to these new disease-modifying drugs. Moreover, more than 10% of
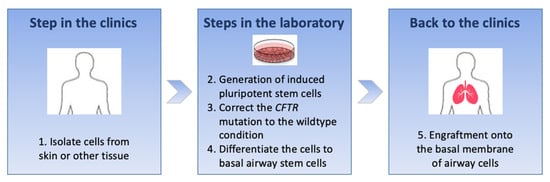
CFTR mutations do not produce any CFTR protein for CFTR modulators to act upon. The purpose of this review is to provide an overview of different approaches pursued to treat patients bearing mutations ineligible for CFTR modulators. One approach is to broaden the numbers of mutations eligible for CFTR modulators. This requires developing strategies to evaluate drugs in populations bearing very rare genotypes. Other approaches aiming at correcting the
CFTR defect develop new mutation-specific or mutation-agnostic therapies for mutations that do not produce a CFTR protein: readthrough agents for nonsense mutations, nucleic acid-based therapies, RNA- or DNA-based, and cell-based therapies. Most of these approaches are in pre-clinical development or, for some of them, early clinical phases. Many hurdles and challenges will have to be solved before they can be safely translated to patients.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
Anti-Inflammatory and Anti-Oxidant Effect of Dimethyl Fumarate in Cystic Fibrosis Bronchial Epithelial Cells
by
Onofrio Laselva, Caterina Allegretta, Sante Di Gioia, Carlo Avolio and Massimo Conese
Cited by 11 | Viewed by 3303
Abstract
Cystic Fibrosis (CF) is caused by mutations on the
CF transmembrane conductance regulator (CFTR) gene and is associated with chronic infection and inflammation. Recently, it has been demonstrated that LPS-induced CFTR dysfunction in airway epithelial cells is due to an early oxidative stress.
[...] Read more.
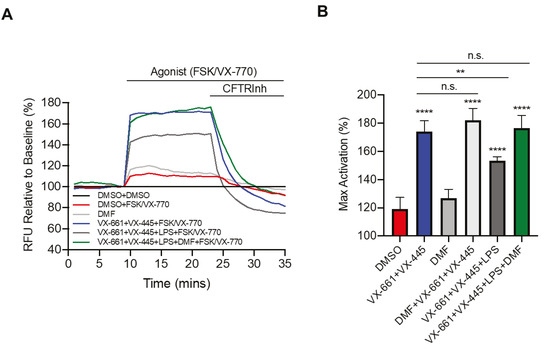
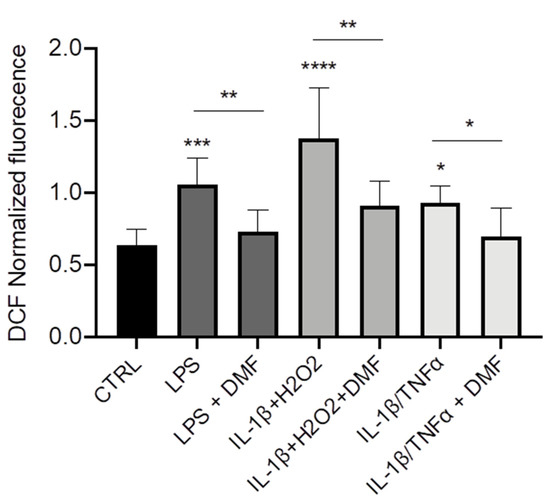
Cystic Fibrosis (CF) is caused by mutations on the
CF transmembrane conductance regulator (CFTR) gene and is associated with chronic infection and inflammation. Recently, it has been demonstrated that LPS-induced CFTR dysfunction in airway epithelial cells is due to an early oxidative stress. Dimethyl fumarate (DMF) is an approved anti-inflammatory and anti-oxidant drug for auto-immune and inflammatory diseases, but its role in the CF has never been investigated. In this study, we examined the effect of DMF on CF-related cytokines expression, ROS measurements and CFTR channel function. We found that DMF reduced the inflammatory response to LPS stimulation in both CF and non-CF bronchial epithelial cells, both as co-treatment and therapy, and restored LPS-mediated decrease of Trikafta™-mediated CFTR function in CF cells bearing the most common mutation, c.1521_1523delCTT (F508del). DMF also inhibited the inflammatory response induced by IL-1β/H
2O
2 and IL-1β/TNFα, mimicking the inflammatory status of CF patients. Finally, we also demonstrated that DMF exhibited an anti-oxidant effect on CF cells after different inflammatory stimulations. Since DMF is an approved drug, it could be further investigated as a novel anti-inflammatory molecule to ameliorate lung inflammation in CF and improve the CFTR modulators efficacy.
Full article
►▼
Show Figures
Planned Papers
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
Title: on CFTR and cell pathophysiology
Authors: Valerie Chappe
Affiliation: Dalhousie University, Physiol & Biophys, HALIFAX, NS, CANADA
Abstract: none
Title: Inflammatory profile of cystic fibrosis bronchial epithelial cells following the recovery of CFTR function: Correctors versus gene expression.
Authors: Mohamed Benharouga; Nadia Alfaidy
Affiliation: In charge of Bachelor-3 Biology and
Master-1 Molecular and Cellular Biology internships
UFR Chemistry/Biology
Faculty of Sciences, Grenoble Alpes University (UGA)
Laboratory: Biology and Biotechnology for Health (BioSanté),
INSERM, U1292
Abstract: none








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}