





Genes 2023, 14(7), 1387; https://doi.org/10.3390/genes14071387 - 1 Jul 2023
Cited by 6 | Viewed by 5016
Abstract
►
Show Figures
Squamates include more than 11,000 extant species of lizards, snakes, and amphisbaenians, and display a dazzling diversity of phenotypes across their over 200-million-year evolutionary history on Earth. Here, we introduce and define squamates (Order Squamata) and review the history and promise of genomic
[...] Read more.
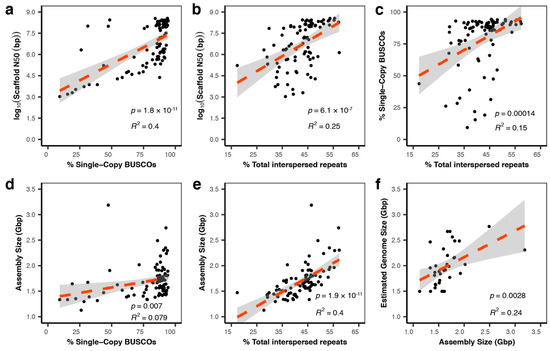
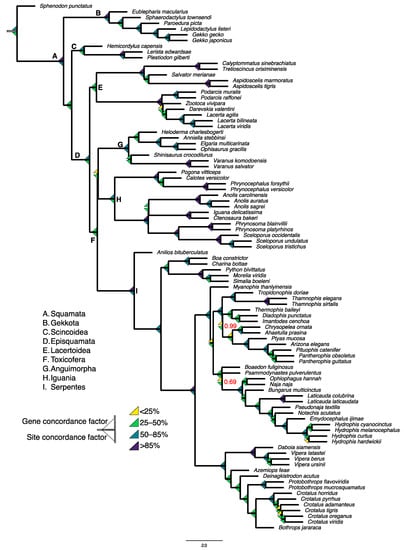
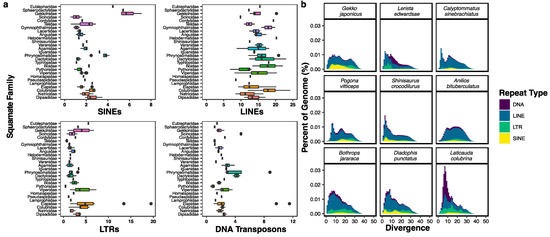
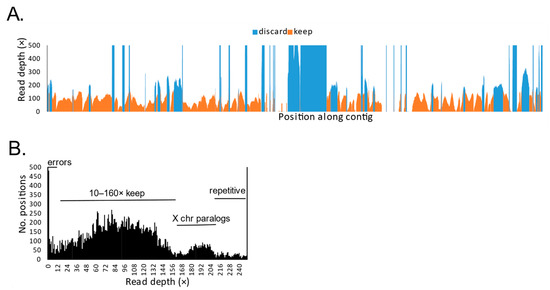
Squamates include more than 11,000 extant species of lizards, snakes, and amphisbaenians, and display a dazzling diversity of phenotypes across their over 200-million-year evolutionary history on Earth. Here, we introduce and define squamates (Order Squamata) and review the history and promise of genomic investigations into the patterns and processes governing squamate evolution, given recent technological advances in DNA sequencing, genome assembly, and evolutionary analysis. We survey the most recently available whole genome assemblies for squamates, including the taxonomic distribution of available squamate genomes, and assess their quality metrics and usefulness for research. We then focus on disagreements in squamate phylogenetic inference, how methods of high-throughput phylogenomics affect these inferences, and demonstrate the promise of whole genomes to settle or sustain persistent phylogenetic arguments for squamates. We review the role transposable elements play in vertebrate evolution, methods of transposable element annotation and analysis, and further demonstrate that through the understanding of the diversity, abundance, and activity of transposable elements in squamate genomes, squamates can be an ideal model for the evolution of genome size and structure in vertebrates. We discuss how squamate genomes can contribute to other areas of biological research such as venom systems, studies of phenotypic evolution, and sex determination. Because they represent more than 30% of the living species of amniote, squamates deserve a genome consortium on par with recent efforts for other amniotes (i.e., mammals and birds) that aim to sequence most of the extant families in a clade.
Full article

Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}