



Int. J. Mol. Sci. 2024, 25(13), 7375; https://doi.org/10.3390/ijms25137375 - 5 Jul 2024
Viewed by 982
Abstract
►
Show Figures
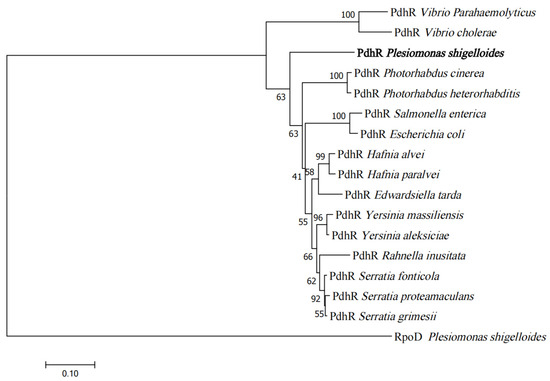
Plesiomonas shigelloides, a Gram-negative bacillus, is the only member of the Enterobacteriaceae family able to produce polar and lateral flagella and cause gastrointestinal and extraintestinal illnesses in humans. The flagellar transcriptional hierarchy of P. shigelloides is currently unknown. In this study, we
[...] Read more.
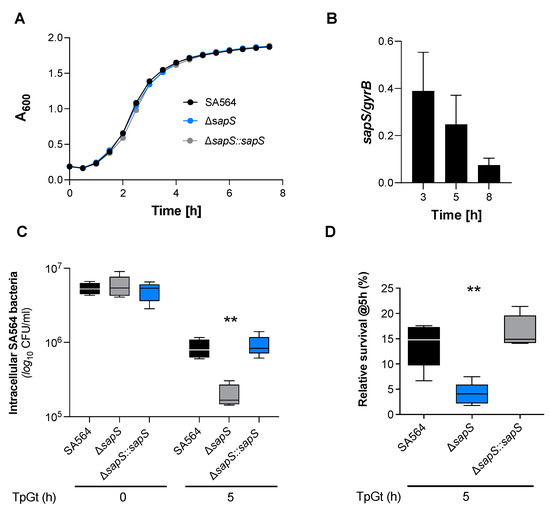
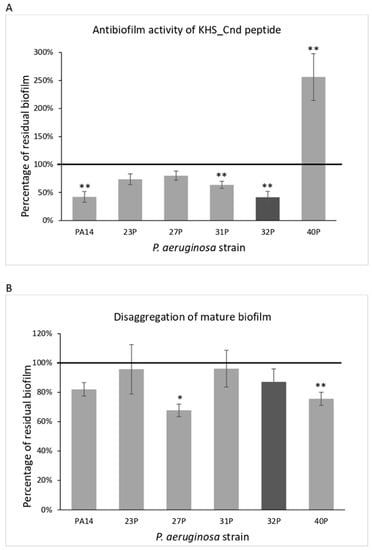
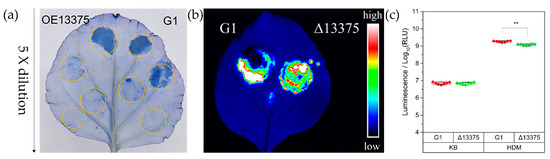
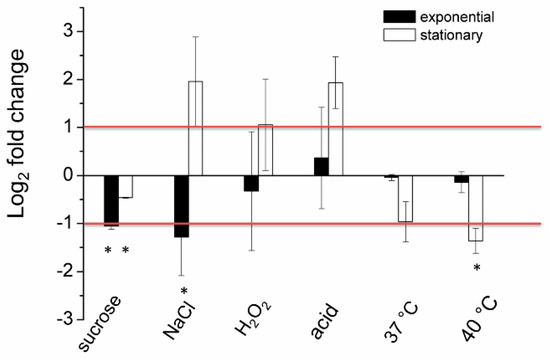
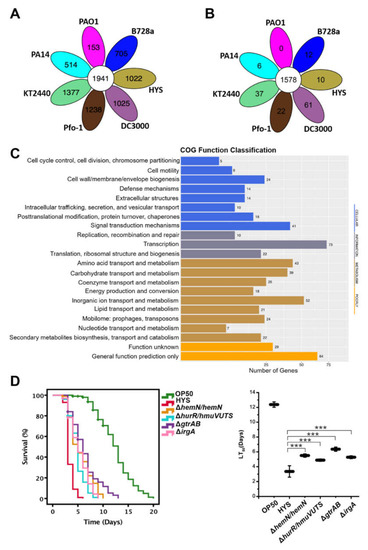
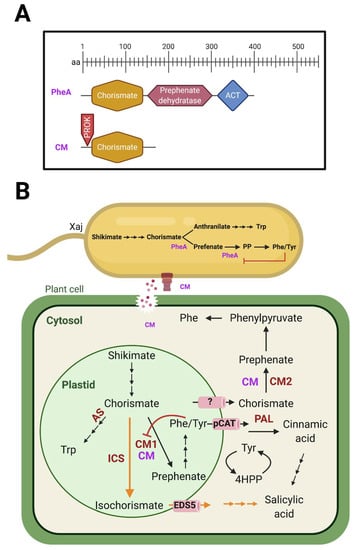
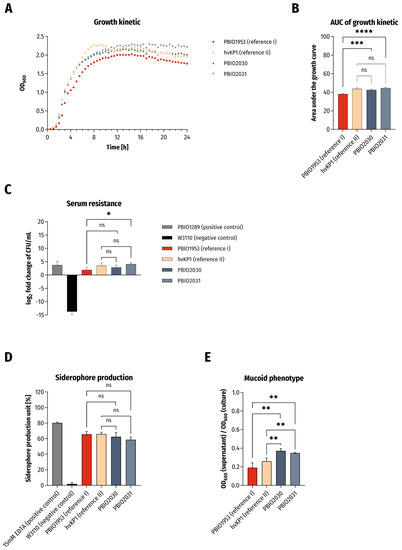
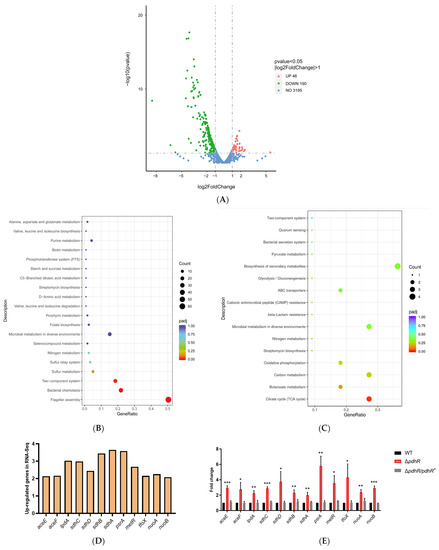
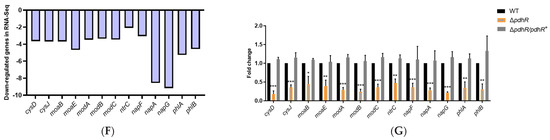
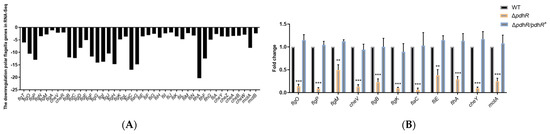
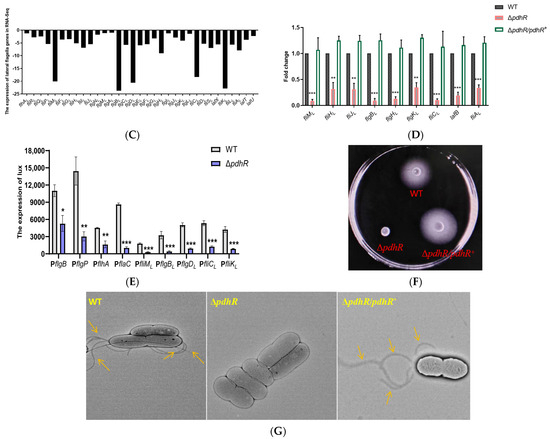
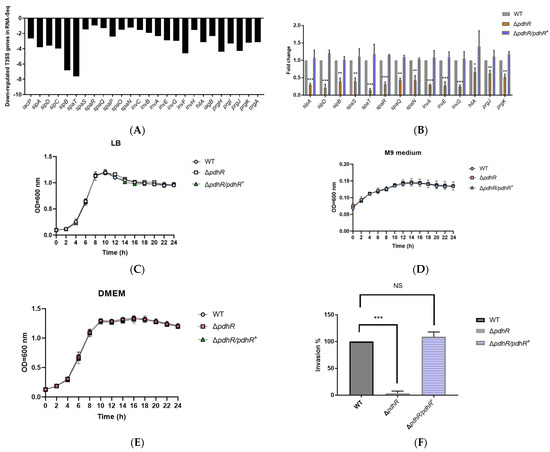
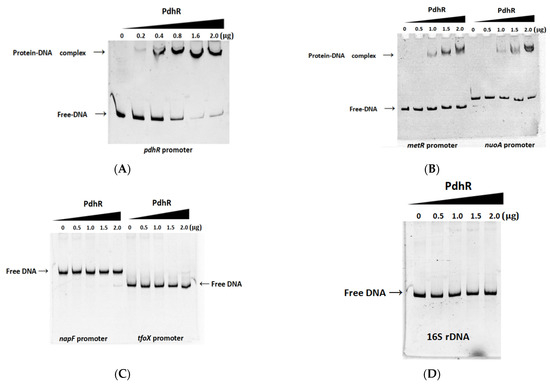
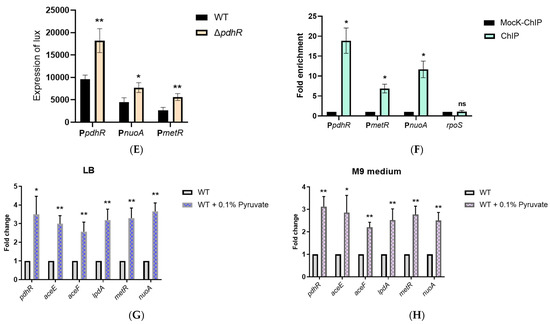
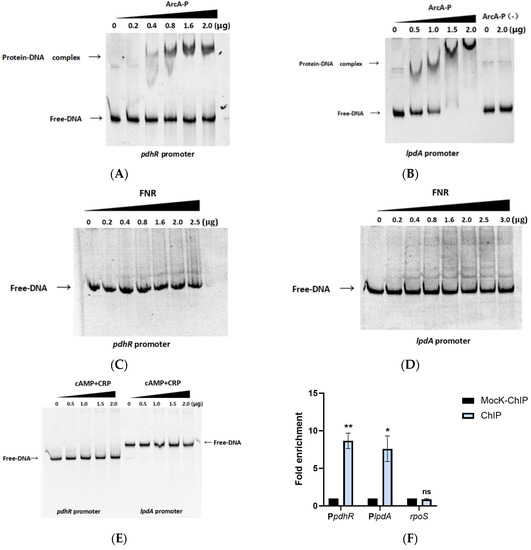
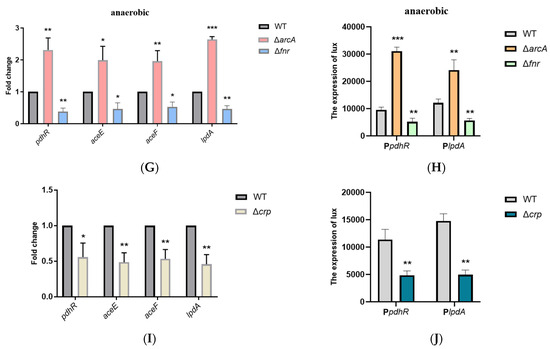
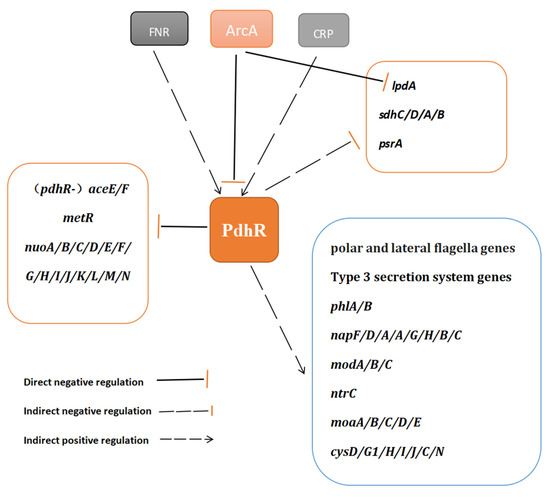
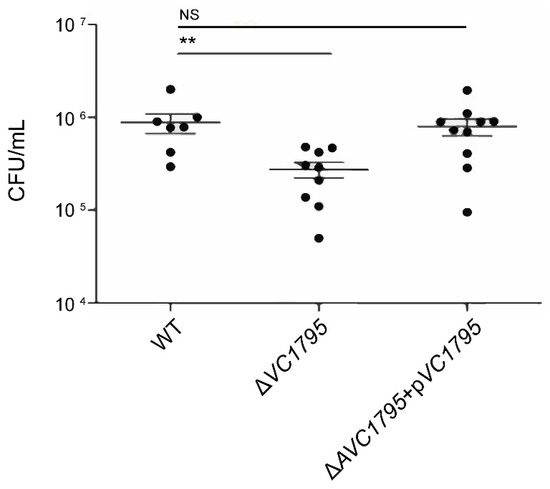
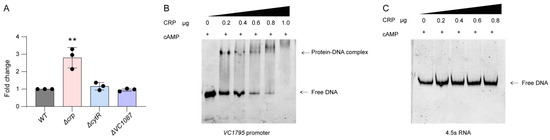
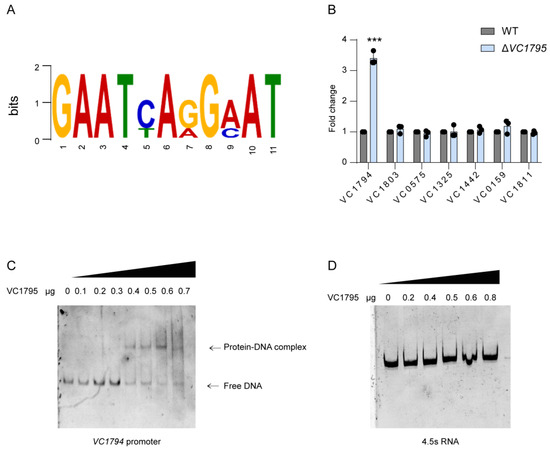
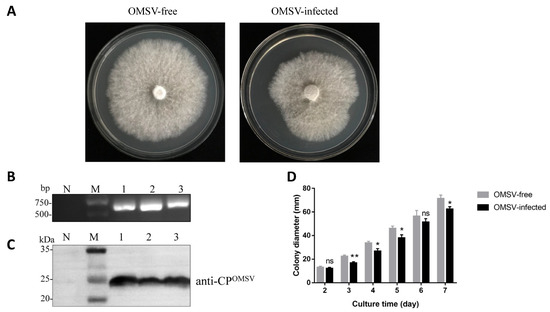
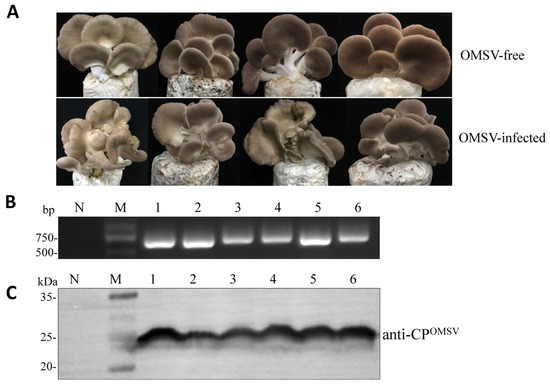
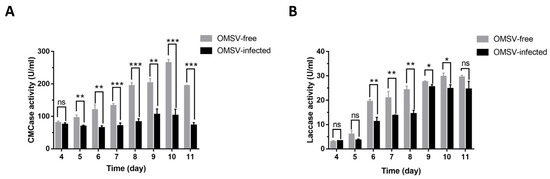
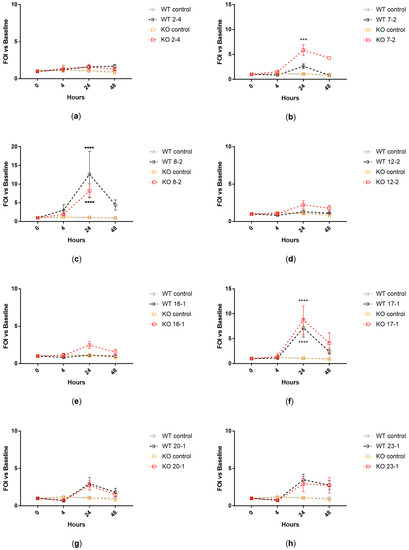
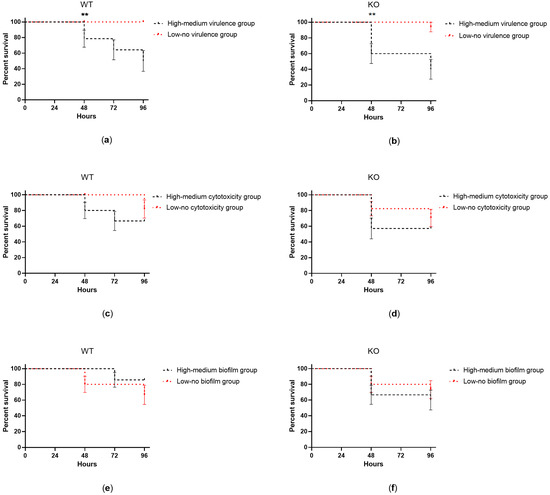
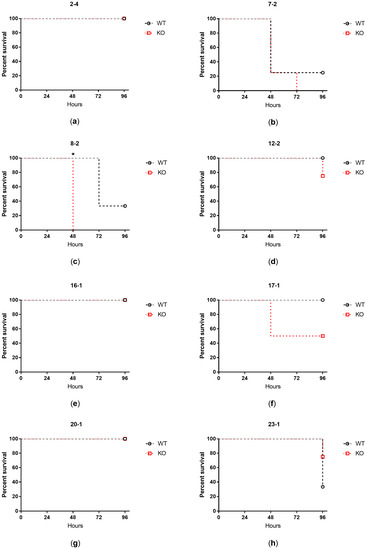
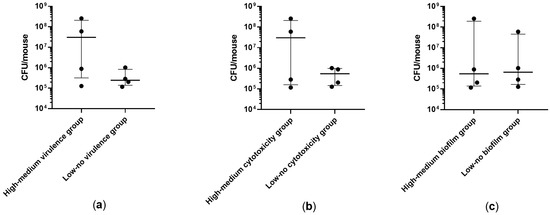
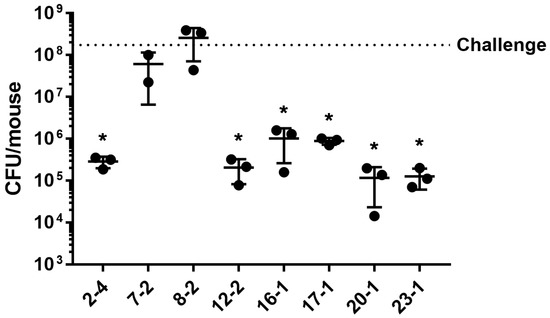
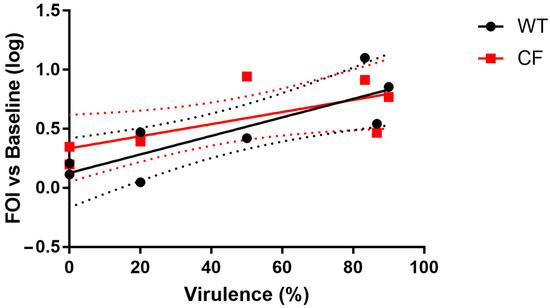
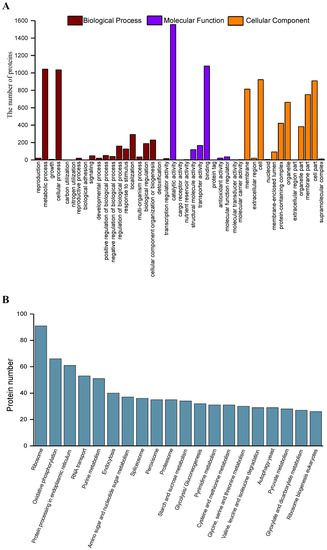
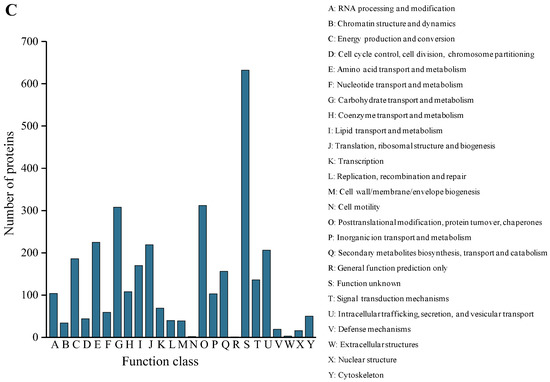
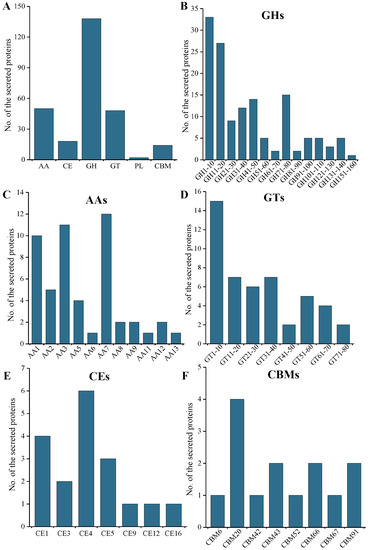
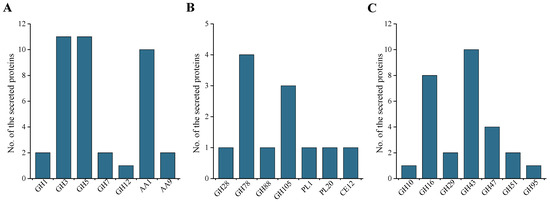
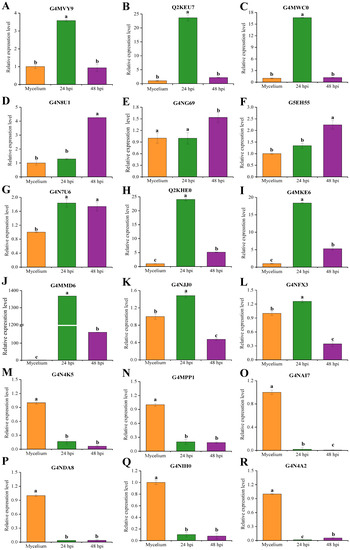
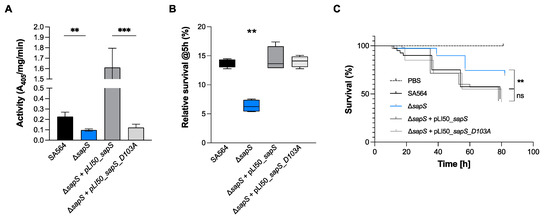
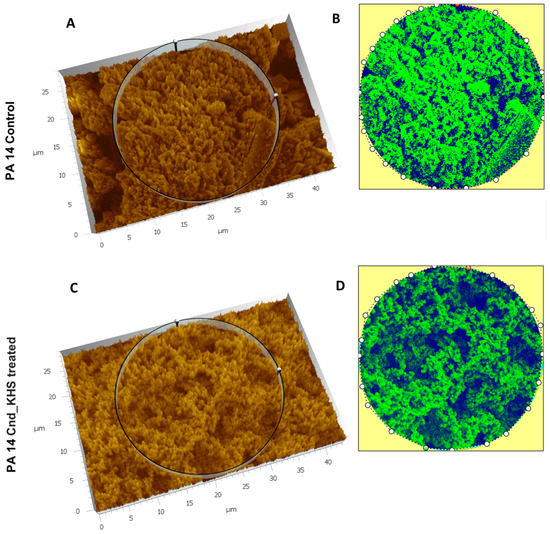
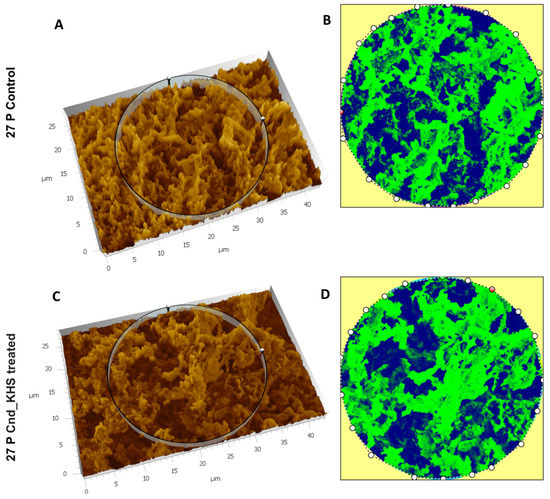
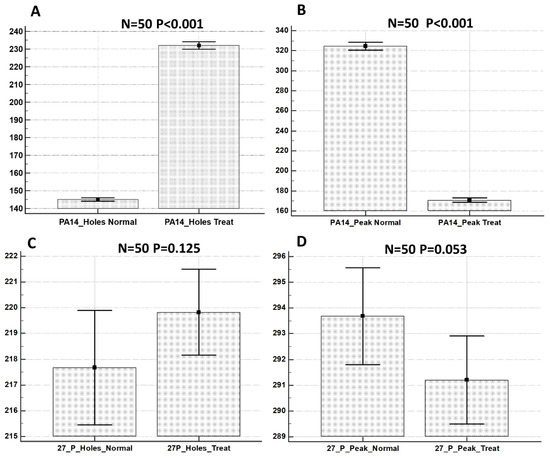
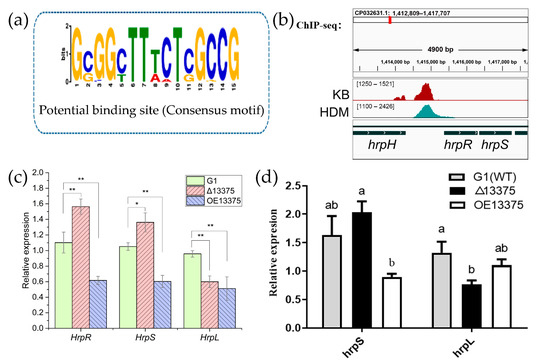
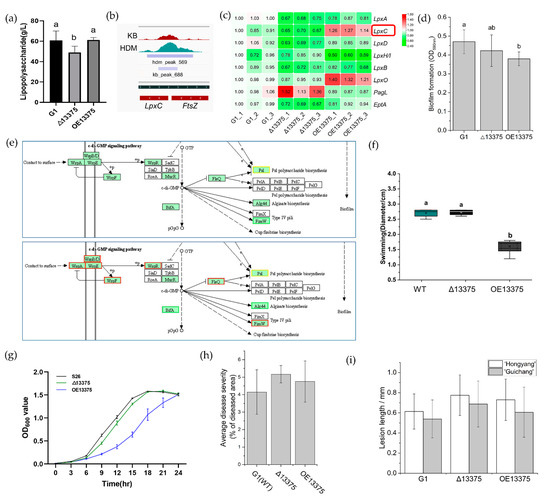
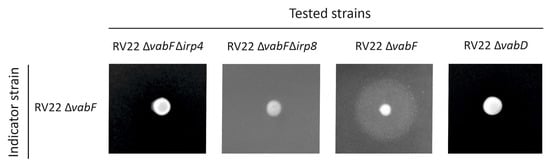
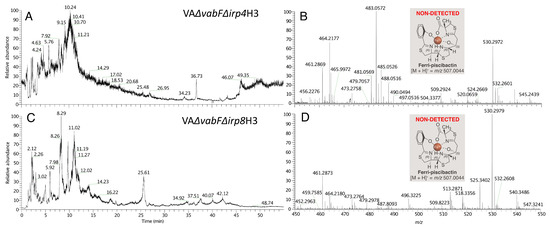
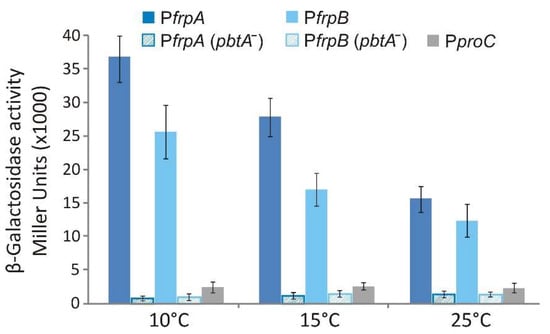
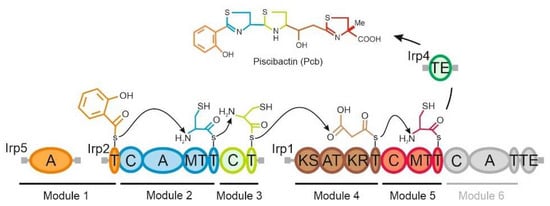
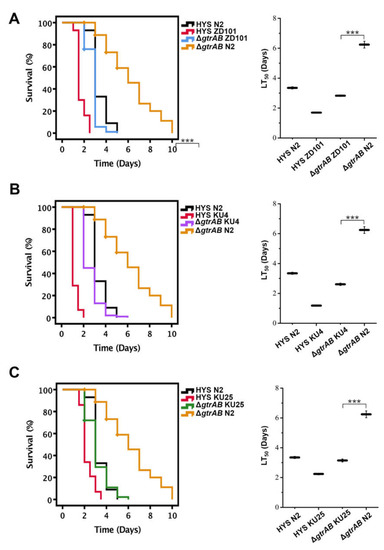
Plesiomonas shigelloides, a Gram-negative bacillus, is the only member of the Enterobacteriaceae family able to produce polar and lateral flagella and cause gastrointestinal and extraintestinal illnesses in humans. The flagellar transcriptional hierarchy of P. shigelloides is currently unknown. In this study, we identified FlaK, FlaM, FliA, and FliAL as the four regulators responsible for polar and lateral flagellar regulation in P. shigelloides. To determine the flagellar transcription hierarchy of P. shigelloides, the transcriptomes of the WT and ΔflaK, ΔflaM, ΔfliA, and ΔfliAL were carried out for comparison in this study. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) and luminescence screening assays were used to validate the RNA-seq results, and the Electrophoretic Mobility Shift Assay (EMSA) results revealed that FlaK can directly bind to the promoters of fliK, fliE, flhA, and cheY, while the FlaM protein can bind directly to the promoters of flgO, flgT, and flgA. Meanwhile, we also observed type VI secretion system (T6SS) and type II secretion system 2 (T2SS-2) genes downregulated in the transcriptome profiles, and the killing assay revealed lower killing abilities for ΔflaK, ΔflaM, ΔfliA, and ΔfliAL compared to the WT, indicating that there was a cross-talk between the flagellar hierarchy system and bacterial secretion system. Invasion assays also showed that ΔflaK, ΔflaM, ΔfliA, and ΔfliAL were less effective in infecting Caco-2 cells than the WT. Additionally, we also found that the loss of flagellar regulators causes the differential expression of some of the physiological metabolic genes of P. shigelloides. Overall, this study aims to reveal the transcriptional hierarchy that controls flagellar gene expression in P. shigelloides, as well as the cross-talk between motility, virulence, and physiological and metabolic activity, laying the groundwork for future research into P. shigelloides’ coordinated survival in the natural environment and the mechanisms that infect the host.
Full article

Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}