1.1. Generalities
This paper presents a review of recent work on time-dependent irreversible processes that differ from more conventional methodologies. It is hoped thereby to provide a more unified approach to topics that usually are treated as disparate subjects.
Thermodynamics is ordinarily applied to systems at equilibrium and subject to reversible processes. However, while considering the Second Law one often confronts the necessity of introducing irreversible processes that follow non-equilibrium paths. This ordinarily requires the introduction of a Taylor series expansion about the quiescent state, to approximate the properties of systems away from equilibrium. Necessarily then, the departures from equilibrium must be “small”. The extent to which such departures can be applied to characterize any situation can only be decided by experiment. In recent times this approach has been extended to encompass more drastic departures from equilibrium, an approach that comes under the heading of extended nonequilibrium thermodynamics.
Here we review a different approach to irreversible processes entirely within the realm of classical thermodynamics, and thus amenable to simple mathematical operations. For a certain class of processes enumerated below we allow the entire system to follow paths well removed from equilibrium.
The present discussion has many historical antecedents. The extension of thermodynamics to nonequilibrium states dates back to Clausius (1850–1865), who first introduced the concept of “uncompensated transformations” for the difference between entropy changes accompanying irreversible as opposed to reversible processes. A similar characterization was developed by Natanson in 1896 [
1]. Jaumann and Lohr (1911–1918) [
2,
3,
4] were among the first to develop a microscopic theory of irreversible thermodynamics, in which they linked the flow of matter, energy, entropy, chemical species and other dissipative processes to the presence of generalized forces. The resulting transport theory leads to fundamental laws discovered earlier through empirical observation: Fourier’s law, Fick’s Law, Ohm’s law, and so on. Later developments, principally by Meixner [
5], de Groot & Mazur [
6], Prigogine [
7], Haase [
8] and their schools then led to the development of phenomenological equations governing several classes of irreversible phenomena. In Extended Irreversible Thermodynamics the assumption of local equilibrium has been abandoned and replaced by the use of fluxes as the fundamental state variables, whence the linear phenomenological equations have been superseded by first order time evolution equations. Thereby memory, nonlinear and nonlocal effects have been successfully incorporated into a coherent description of such nonlinear phenomena. Detailed expositions are provided in References [
9,
10,
11,
12,
13,
14].
The subsequent discussion has its origins in the work of De Donder [
15] who related “non-compensated heat” in a chemical reaction directly to the corresponding affinity function. Later work on time-dependent irreversible processes has been reviewed by Bejan [
16] and was brought up to date in a review by Lucia and Grazzini [
17,
18]. As emphasized in both publications, time must be introduced as an important variable in the specification of irreversible processes. This view has been adopted in the present approach, with emphasis placed on the time evolution of irreversible processes rather than on setting up time-dependent differential equations and optimization of available work in engineering applications, which has been the goal of the majority of publications in the area. The publications that come closest to the present approach are probably those of Salamon, Band, and Kafri [
19], Band, Kafri, and Salamon, [
20], Ondrechen, Rubin, and Band [
21], who sketched the use of time as a parameter in specifying the course of an irreversible process, again, with an emphasis on optimization of available work.
1.2. Basic Assumptions
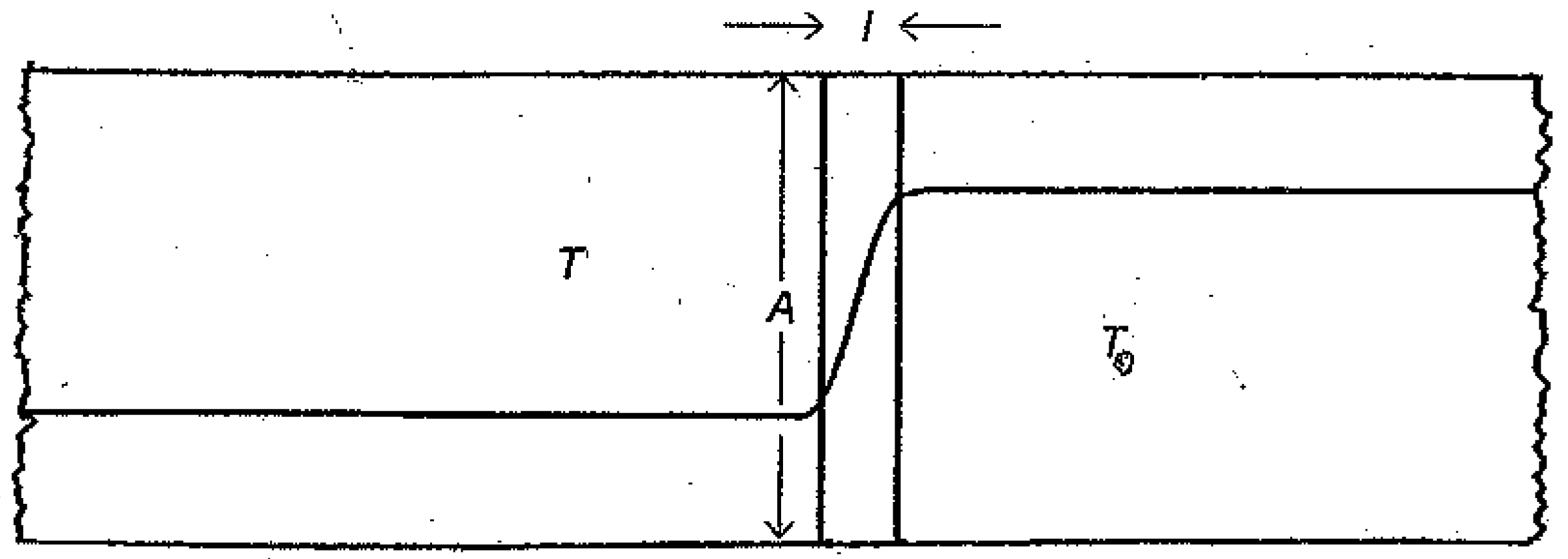
We now consider an alternative approach to irreversible processes by examining a system in contact with a reservoir, the compound unit being isolated from the rest of the universe. A sketch of the isolated unit is shown in
Figure 1. The reservoir is assumed to be large in extent and maintained well mixed so that any change in its configuration occurs reversibly while maintaining all intensive variables constant, even in its interactions with the system undergoing irreversible changes. This assumption, commonly employed, is crucial, so as to maintain a simple mathematical framework for later developments. The reservoir is linked to the system via an intervening moveable junction that is a poor thermal conductor, also allowing for the slow diffusion of chemical species between the compartments. As indicated in the diagram, the temperature
T, pressure
P, and chemical potential
μ of the system differ from the corresponding
T0,
P0,
μ0 of the reservoir. During the interaction the system is assumed to undergo slow physical and chemical changes, so as to allow for a uniform change in all its properties. This process will be designated as a
quasistatic irreversible process (QSIP). Thus, while the temperature
T0 of the reservoir remains fixed, the temperature
T of the system is forced to change sufficiently slowly so as to keep
T essentially uniform over almost the entire volume of the system. The temperature change then occurs solely within the boundary region. Similar considerations apply to other intensive variables. The two compartments are thus not in equilibrium with each other; instead, one can maintain arbitrarily large differences in intensive variables. We specify the resulting thermodynamic relations under such conditions and determine the operating characteristics in terms of thermodynamic equilibrium variables. These idealizations may be approached by choosing a system that has a fast relaxation time, and is small in the lateral extension away from the junction. Alternatively, one may consider a set of small subsystems dispersed within the reservoir that undergo identical changes simultaneously.
Figure 1.
Temperature Profile for a System at Temperature T Attached to a Reservoir at Temperature T0 via a Junction of Cross Section A and Length l.
Figure 1.
Temperature Profile for a System at Temperature T Attached to a Reservoir at Temperature T0 via a Junction of Cross Section A and Length l.
Under the above assumptions the temperature of relevance is always that of the reservoir and is thus well defined. If one wishes to relax the assumption of a uniform temperature distribution in the system one needs to consider the extent to which the concept of temperature remains applicable for large departures of the system from equilibrium. This matter has been addressed in several studies [
22,
23,
24], where it is shown that the concept of temperature remains relevant and does not depart significantly from the temperature of the applicable equilibrium state. This in principle allows one to investigate cases where the temperature of the system is no longer uniform. The resulting complications are not addressed here.
1.4. Entropy Changes during Processes
We begin a study of entropy changes of QSIPs in the isolated compound system. Consider a system that in its interactions with a reservoir undergoes a reversible change from state A to state B, at a uniform temperature
T. The entropy change of the compound system in an incremental step is specified by:
where conventional notation was employed; the zero subscript refers to the reservoir. Now let the same step be executed irreversibly, with the two components at different temperatures; normally this is recorded via the inequality:
To eliminate the inequality we introduce a deficit function, commonly known as the entropy dissipation function,
đθ > 0, such that:
So far
đθ is simply a bookkeeping device, but it acquires meaning when we subtract Equation (1) from Equation (3) to write:
whence
đθ represents the difference in entropy change of the surroundings when the system is allowed to undergo the same process irreversibly as opposed to reversibly.
In the isolated compound unit all of the heat absorbed (released) by the system is transferred out of (into) the reservoir. Thus, during the reversible (
r) operation the entropy changes of the system and surroundings are related by:
while the irreversible (
i) operation entails an entropy change:
where we invoked the reversibility of all processes in the reservoir. On inserting Equations (5) and (6) into Equation (3) we obtain a relation between heat transfers during the reversible and irreversible operations:
or in alternative form as:
Equation (7) duplicates the expression derived by Tolman and Fine (1948) [
25] by a different method.
For heat flow into the system
điQ and
đrQ are both positive and
T < T0. The inequality in Equation (7) must hold even for the limiting case
T → T0−. This leads directly to the inequality set:
which represents one statement of the
Clausius inequality. For the reverse flow both
điQ and
đrQ are negative and
T > T0; then Equation (9) represents a string of increasingly less negative quantities.
Equation (7) may be turned around to read:
which shows explicitly that in irreversible processes the total entropy change in the system is only partially accounted for by the transfer of heat. Note that it is the well established temperature of the reservoir that enters the expression. For adiabatic processes in isolation,
δS = đθ > 0; entropy can only increase, a process that continues until quiescence is reached, at which stage the entropy is at a maximum with respect to the applied constraints. All this obviously is in accord with well-established principles.
If we apply the
điQ < đrQ inequality to Equation (8) we require the last two terms on the right to be negative, so that:
where the lower bound is always positive, since for
T/T0 < 1, đrQ > 0, and for
T/T0 > 1,
đrQ < 0. Thus, Equation (11) yields a lower positive bound on
đθ.
The performance of work
W may be analyzed on the basis of the First Law in the formulation:
On substituting from Equations (7) or (8) we find that:
Equation (13) represents a variant of the
Gouy-Stodola theorem.
Comparison with Equations (8) and (11) shows that điW > đrW, which is another well established principle.
We may solve Equation (13) for:
This finally provides one method for determining đθ through measurements of work performance for the same process under actual as opposed to reversible operating conditions; also needed are measurements or calculations of the entropy change under those conditions.
In the absence of work performance Equation (14) reduces to:
which was derived elsewhere by Kestin by different means [
26]. The first term in Equation (14) was obtained Bejan who employed a different approach [
27,
28]. Equation (14) further provides a thermodynamic background for the theoretical work of Jarzynski [
29] who related the average of repeated executions of irreversible microscopic processes to thermodynamic equilibrium processes. That work was confined to processes executed at constant temperature; the above relation shows how this analysis may be extended.
We next set up functions of state that incorporate irreversible processes; these can then be used to specify đθ for processes under a variety of operating conditions.
1.5. The Energy
We begin by applying the First Law to the operation of the
reservoir, in its interactions with the system that involve heat exchange, mechanical work, and transfer of matter. For the corresponding reversible operation we write in conventional notation:
for the energy of the surroundings. The energy of the
system is found by noting that in the isolated compound unit
dE + dE0 = 0. Further, when the systems undergoes irreversible processes we use Equation (3) to replace
dS0 in Equation (16) by
dbS0 = −
dS + đθ. On the assumption that the isolated compound system is maintained at constant volume and fixed composition we may also set
dV0 = −
dV, and
dn0i = −
dni, so that the energy differential of the
system is given as:
This relation, involving different arguments, was derived by Kestin [
26]. The intensive variables are those of the surroundings and are therefore well defined, even under irreversible processes within the system;
dS,
dV,
dni are the control variables;
đθ may be specified by Equation (14), or by expressions developed shortly, or by techniques reviewed below. Thus,
dE is well defined for QSIPs. If work other than mechanical is involved, relevant terms must be added to Equation (17) as a product of intensive variables appropriate to the relevant work reservoir, and corresponding extensive variables for the system proper.
It is instructive to rewrite Equation (17) in the form:
where the variables lacking the subscript zero refer to the properties of the system. Under reversible operations the first three terms and the last term drop out, and one recovers the conventional formulation. Since
E is a function of state we next invoke the relation
dE = TdS − PdV. + Σ
iμidni for reversible operations, and subtract this relation from Equation (18), which allows us to solve for:
The quantities on the right are experimentally accessible; hence, the above equation permits the determination of đθ in terms of S, V, and ni as the control variables. The above relation is not immediately useful since the specification of dE involves the use of the state function dS as a control variable. This problem is addressed as follows:
1.6. The Helmholtz Free Energy
To construct the Helmholtz energy we introduce the definition
A =
E –
TS, which sensibly involves the temperature
T of the system. This transforms the independent variable from
S to
T. Based on Equation (18) we find that:
This must be rewritten in terms of
T, V, ni as the applicable control variables. For this purpose we now express entropy in terms of these quantities:
S = S(T,V,ni), with the differential form:
Substitution in Equation (20) then yields:
We now introduce the replacements (∂S/∂T)V,ni = CV/T, and (∂S/∂V)T,ni = (∂P/∂T)V,ni, where CV is the heat capacity at constant volume and composition, and where the appropriate Maxwell relation has been introduced.
Also, we use the mathematical identity
and then replace the numerator and denominator by -
αV and by -
βV, where
α and
β are the isobaric coefficient of expansion and the isothermal compressibility respectively. We also set
as the differential entropy at constant temperature, volume and mole number of species
j ≠ i. Then:
which relates the differential of the Helmholtz function under non-equilibrium conditions to changes in
T,
V, and the
ni. The associated coefficients involve measurable quantities. Again, under reversible operations
dA = - SdT - PdV + Σ
iμidn; when this relation is subtracted from (23) we may solve for:
which specifies
đθ in terms of
T, V, and
ni; all the coefficients involve measurable quantities.
1.7. The Gibbs Free Energy
Using by now familiar methodology, we define the Gibbs energy by the relation
G = E – TS + PV. When converted to differential form we write:
for use under non-equilibrium conditions. To introduce the proper control variables we express the entropy function as
S = S(T,P,ni) and the volume as
V = V(T,P,ni), with corresponding differentials for
dS and
dV. On inserting these in Equation (25) we obtain:
We next set (∂
S/∂
T)
P,ni = CP/T, introduce the Maxwell relation (∂
S/∂
P)
T,ni = - (∂
V/∂
T)
P,ni and
. replace the partial derivatives - (∂
V/∂
P)
T,ni and (∂
V/∂
T)
P,ni by
βV and by
αV respectively. Here (
∂S/∂
ni)
T,P,ni≠j and (
∂V/∂
ni)
T,P,ni≠j represent partial molal entropies
![Entropy 15 02975 i002]()
and volumes
![Entropy 15 02975 i003]()
. We then rewrite Equation (26) in the less unwieldy form:
for the Gibbs function under non-equilibrium conditions. Then subtract the standard reversible form
dG = –
SdT + VdP + Σ
iμidni and solve for:
which expresses
đθ in terms of
T, P, and composition.
1.8. The Enthalpy
Lastly, we turn to the enthalpy
H = E + PV. By the customary technique we develop the differential form
dH = dE + PdV + VdP and then insert Equation (18) to obtain:
Here
S, P, and composition are the applicable control variables. We therefore consider the volume first in the form
V = V(
P,T,ni)
. We next introduce the entropy as a function of the same variables:
S = S(
P,T.ni)
, which function we invert to read
T = T(
S,P,ni)
. Lastly, we insert this expression into the equation of state: thus,
V = V(
P,T(
S,P,ni)
,ni)
≡ V(
S,P,ni)
. Then:
Now we introduce Equation (30) into Equation (29) to obtain:
for the infinitesimal enthalpy change under nonequilibrium conditions. Finally, subtract the standard reversible form
dH = TdS + VdP + Σ
iμidni and solve for:
which yields the deficit function in terms of
S,
P, and
ni.
This completes the task of specifying đθ in terms of the applicable control variables.

{kind=link}

 dni] – (P0 – P)dV + Σi(μ0i – μi)dni – SdT – PdV + Σiμidni – T0đθ
dni] – (P0 – P)dV + Σi(μ0i – μi)dni – SdT – PdV + Σiμidni – T0đθ
 and volumes
and volumes  . We then rewrite Equation (26) in the less unwieldy form:
. We then rewrite Equation (26) in the less unwieldy form:
 + (μ0/T0) (1 – μ /μ0)]dn = (1 – T/ T0)[(CV/T)dT ] + [
+ (μ0/T0) (1 – μ /μ0)]dn = (1 – T/ T0)[(CV/T)dT ] + [  /T0]dn
/T0]dn



