A Study of Fractality and Long-Range Order in the Distribution of Transposable Elements in Eukaryotic Genomes Using the Scaling Properties of Block Entropy and Box-Counting

Abstract
: Repeats or Transposable Elements (TEs) are highly repeated sequence stretches, present in virtually all eukaryotic genomes. We explore the distribution of representative TEs from all major classes in entire chromosomes across various organisms. We employ two complementary approaches, the scaling of block entropy and box-counting. Both converge to the conclusion that well-developed fractality is typical of small genomes while in large genomes it appears sporadically and in some cases is rudimentary. The human genome is particularly prone to develop this pattern, as TE chromosomal distributions therein are often highly clustered and inhomogeneous. Comparing with previous works, where occurrence of power-law-like size distributions in inter-repeat distances is studied, we conclude that fractality in entire chromosomes is a more stringent (thus less often encountered) condition. We have formulated a simple evolutionary scenario for the genomic dynamics of TEs, which may account for their fractal distribution in real genomes. The observed fractality and long-range properties of TE genomic distributions have probably contributed to the formation of the “fractal globule”, a model for the confined chromatin organization of the eukaryotic nucleus proposed on the basis of experimental evidence.1. Introduction
In information theory, the notion of entropy was conceived by Shannon [1] to estimate the amount of information that is carried in a transmitted message. During the last decades, scale invariance and fractality have been found in time series from signal transmission in electronic engineering, earthquakes, economy, social sciences and many other fields. Very often, such studies have been carried out using the standard box-counting technique and in several cases of systems characterized by long range correlations Shannon entropy has been used.
In a previous work [2] we studied the scaling properties of the block entropy of the distribution of genes in eukaryotic genomes through a coarse-graining reduction of the DNA sequence into a symbol sequence. The convention that we followed was that zeros “0” in the symbol sequence stood for non-protein-coding nucleotides and ones “1” for nucleotides belonging to protein coding segments (exons). Several studies have shown that a linear scaling of the Shannon-like (or block) entropy H(n) with the length n of the word (called hereafter n-word) in semi-logarithmic plots is a clear indication of long-range order and fractality, as we are going to discuss in the next section [3–6]. We verified this conjecture numerically in the case of finite Cantor-like symbol sequences [2]. Then, we showed that the genomic distribution of protein coding segments often exhibits this particular scaling.
Transposable Elements (TEs), also termed (interspersed) repeats, are sequence segments which are present in virtually all eukaryotic genomes in many (often several thousands) copies per chromosome [7–9]. Two major types of TEs have been distinguished on the basis of their means of genomic proliferation [8]. Retrostransposons proliferate through the intermediate of a messenger RNA sequence, which is subsequently reverse transcribed to a DNA copy, this last being randomly integrated back into the host genome. The other major class of TEs, DNA transposons, do not go through RNA intermediates during their self-replication. In previous works [10,11] we have shown, by studying the size distribution of inter-repeat distances, that the spatial arrangement of all principal classes of TEs in 14 representative genomes from phylogenetically distant organisms exhibit long-range correlations. Often, these distributions are power-law-like, with their linear region in double logarithmic scale extending up to three orders of magnitude.
We here study the distribution of several specific types of repeats in the following way: Nucleotides of the chromosome are replaced by zeros, if they do not belong to the repeat type under consideration and by ones if they belong to it. Thus, the juxtaposition of short islands formed by ones interrupting the continuum of zeros reflects the pattern of the spatial chromosomal arrangement of this specific repeat type. Then we proceed with the generated binary symbol sequence as follows: (i) The scaling of the block entropy H(n) vs. n in this symbol sequence is examined and quantified by means of the extent of linearity in semi-logarithmic plots; and (ii) A box-counting method is applied and the similarity dimension is computed along with the extent of linearity in double logarithmic scale.
The content of this article is organised as follows: in the Methods section we describe the block entropy scaling and box-counting methodology. We then list the sources of the genomic data used and describe the Insertion-Elimination model. In the Results section, the two methods described previously are applied to the genomic coordinates of selected repeat (TE) populations. In the Discussion section we elaborate on the compatibility of these results with the proposed model and, more generally, on their significance and implications for genomic evolution. In the final section the main conclusions of this work are briefly recapitulated.
2. Methods
2.1. Block Entropy Scaling
Let us suppose a symbol sequence of length N, with symbols taken from a binary alphabet {0, 1} and let pn(A1,..., An) be the probability to find the block or n-word (A1,..., An) in this sequence. The Shannon-like entropy for n-words, or block entropy, is defined as [12]:
H(n) can be interpreted as a measure of the mean uncertainty of the prediction of an n-word. A standard treatment and description of the essential properties of block entropy and of other related quantities may be found in [3–5]. Here we briefly summarize only some essential results with immediate relevance to the purposes of the present study, while further analysis and more details can be found in [2].
In the literature one can meet two ways of reading the symbol sequence and extracting the probability distribution of n-words; by “gliding” and by “lumping”. Throughout this work, symbol-sequences are read by “lumping”. This means that, instead of exhaustively reading all possible words of length n (gliding), only n-words sampled with a constant step equal to n are considered. Equivalently, we can say that after reading the initial n-word of the sequence, the next counted n-word is the one starting at n + 1 and so on up to the end of the sequence. Thus, each letter of the sequence belongs only to one counted n-word.
The scaling properties of the block entropy have been used as a measure for the classification of the symbol sequences. Crucial scaling features of H(n) have been investigated by several authors. Ebeling and Nicolis [4] have conjectured the following specific form for the scaling of H(n):
for symbolic sequences generated by non-linear dynamics including language-like processes [4–6]. More specifically, in the case of the Feigenbaum attractor of the logistic map and for n = 2k (k = 2, 3, 4 …), Grassberger [3], see also [12,13], has shown that for reading the sequence by gliding, the following scaling holds:
In this system linearity in semi-logarithmic plot holds (see [14] and references given therein), which in terms of Equation (2) corresponds to: g ≠ 0, h = 0, μ0 = 0, μ1 > 0 [15]. This type of scaling is conjectured to hold for a large class of symbol-sequences with fractal properties. Thus the H(n) – logn linearity is related to the scale-free structure of such sequences entailing the existence of long-range correlations. We have previously verified this conjecture for both deterministic Cantor-like symbol-sequences and probabilistic ones, which present features closer to genomic sequences [2].
For all genomic and simulated data sets we generated surrogate random sequences with the same 0/1 composition and lacking, by construction, any internal structure. Specifically, we reconstructed a sequence with the same length as the genomic one by spreading the biological functional units (repeats) in random positions. The curves showing the entropic scaling (H(n) vs. n) of the original sequence and of its surrogate are presented in the same plot.
We quantify the fractality of a considered sequence by the extent of linearity in the semi-logaritmic scale E and the corresponding slope S. When more than one linear segment exists, we denote with E* the sum of their lengths. One additional quantity we introduce here and use heuristically as an estimator of the degree of organization of a sequence is the ratio R of the entropy value of the surrogate sequence over the entropy value of the studied (genomic or simulated) sequence. This ratio is always calculated for the value of n where the surrogate sequence presents its maximum entropy value, before the finite size effect completely distorts its shape. High values of R denote a high degree of order – and possible fractality – of the studied sequence.
We introduce a shrinkage factor (s.f.) allowing a compression of the genomic sequence. For s.f. equal to e.g., ten symbols, we start from the beginning of the chromosome and we substitute every ten “0” by one “0” and every ten “1” by one “1”. When we meet a 10-letter string of mixed composition we substitute it by a single “1”. We choose to present our results for s.f. = 10, while in Section 1 of the supplementary material, the entropy scaling for various s.f. values along with the case of no shrinkage are presented. We have verified that shrinkage does not alter our results. Further details on the use of s.f. and on the inclusion in the presented plots of n-words with large values of n can be found in [2].
2.2. Box-Counting Method for Estimating the Extent of Fractality and Fractal Dimension
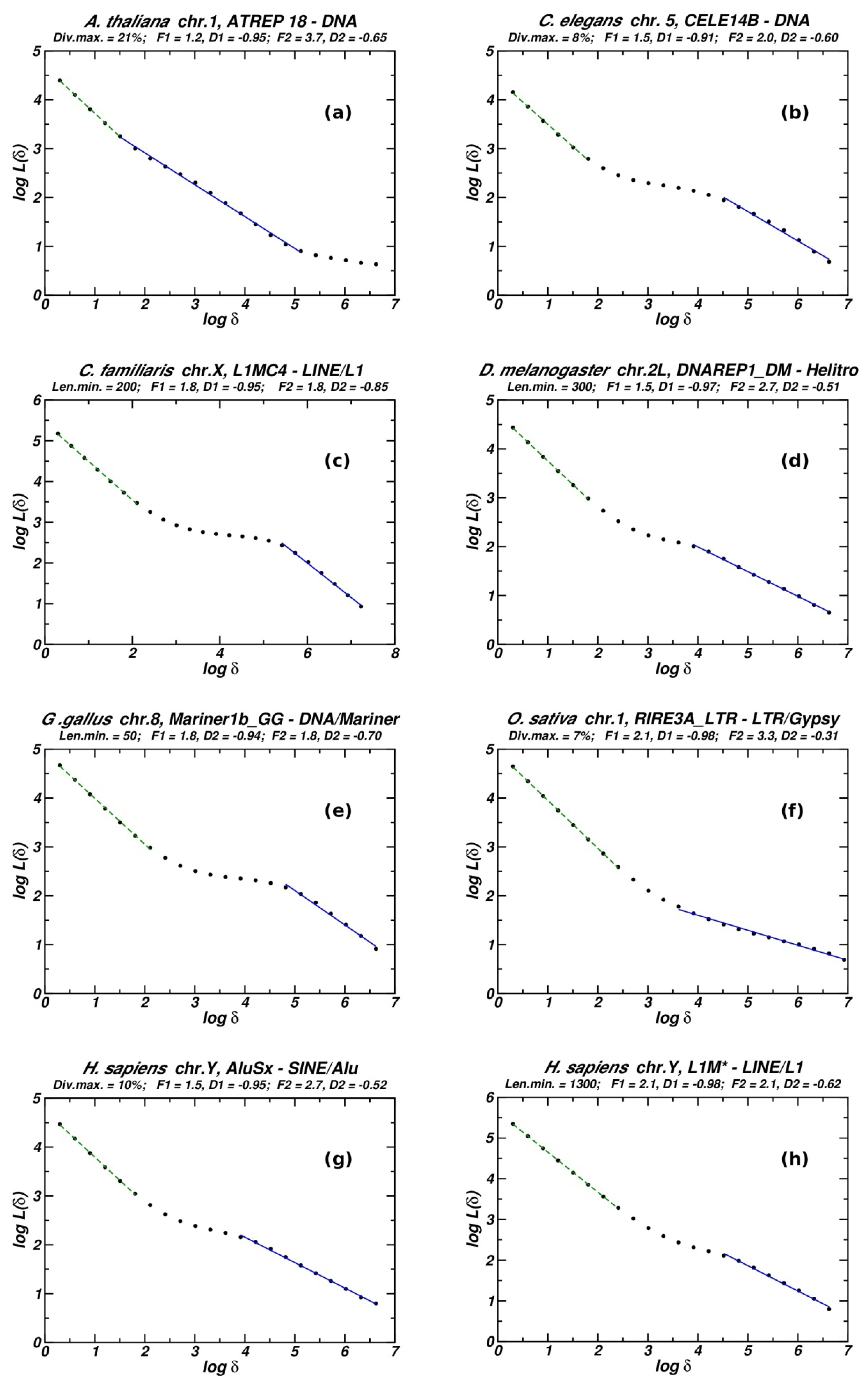
Box-counting is a classical method for estimating the fractal characteristics in a set of data [16,17]. We use a simple one-dimension implementation covering the chromosomal length by one dimensional “boxes” of length δ. The number of such boxes overlapping (fully or partially) at least one repeat copy is considered to represent the chromosomal length L(δ) occupied by TEs. When fractality holds, the measured length shows no sign of reaching a fixed value as δ decreases [17]. The measured length scales as, L(δ) ~ δD, with the exponent D being the negative fractal dimension Df of the studied object. The plots included herein, which show how L(δ) scales as a function of δ, are presented in a double logarithmic scale. We are interested in both the slope of the linear part of the curve and the extent of the linearity. Here fractality is considered to hold if Df takes values lower that 0.9 for a linear extent (F) exceeding one order of magnitude. The values at the limits of the linear region determine the lower and upper cutoffs, between which the studied spatial pattern is, statistically, self-similar. We compute L(δ) ten times, for each value of δ, with a frame shift equal to 1/40 of the total sequence length and then, we average in order to obtain results independent of the choice of the starting point of the measurement.
Notice that for a genomic TE distribution two linearity regions are observed in most cases: one in the low-length region related to the length of the studied TEs and one in the high-length region. The latter is the only one for which the slope is found to significantly deviate from −1 (in the cases showing fractality).
2.3. Genomic Sequences Retrieval and Repeat-Coordinates Extraction
Our analysis requires a large sample of TEs for each distribution, thus in this study we have included only TE populations with relatively large copy number per chromosome. We have selected chromosomes from several taxonomically distant species.
The genomic coordinates of TEs were extracted from RepeatMasker output files downloaded from the University of California Santa Cruz (UCSC) genome browser [18], with the exception of C. elegans for which the assembled chromosomes (release on 9 November 1998) were downloaded from the National Center for Biotechnology Information (NCBI) Genomic Biology [19].
In all cases the repeat annotation was performed by a standard program that screens DNA sequences for interspersed repeats and low complexity DNA sequences (RepeatMasker) [20] with the –s (sensitive) setting, using libraries from the most commonly used database of repetitive DNA elements (RepBase) [21] and the Washington University Basic Local Alignment Search Tool WU-BLAST [22], which finds regions of sequence similarity, as search engine.
The data for the repeat populations of the studied genomes were extracted after a suitable parsing of the standard RepeatMasker output. A maximum divergence (Divmax) is set as an upper limit throughout our main collection of repeats studied in this work (see Table 1) in order to select a repeat sub-population with limited dissimilarity with the family consensus. Thus, only the nucleotides of these repeat-copies are replaced by ones, while the rest of the nucleotides of a studied chromosome are denoted by zeros. We use the divergence computed by the RepeatMasker, defined as: “% substitutions in matching region compared to the consensus” [20]. For transposable elements with a strong tendency to generate severely truncated copies (e.g., L1s) a minimum length (Lenmin) limit has been used analogously. Additionally, some cases of full repeat populations are also presented, in order to show the effect of heavily deteriorated TE copies on the observed fractal pattern. The reasons for a maximum divergence (or minimum length) dependent treatment is presented in detail in [10,11] and discussed herein, later on, in relation to the proposed model. The 33 cases of eukaryotic chromosomes we chose to study, exhibit power-law-like distributions of the inter-repeat distances (data from references [10,11]). When chromosomes lacking this feature are examined, box-counting and entropic scaling invariably fail to identify any indication of fractality (figures not shown).
2.4. The “Insertion-Elimination Model”
In previous works we formulated an evolutionary model describing genomic dynamics relevant to TE evolution [10,11]. We here briefly describe the structure of the model and refer the reader to these previous works for a detailed description of the biological background. The insertion-elimination model for TE genomic dynamics builds upon models for the explanation of fractality in aggregation patterns in physicochemical systems [23]. Our model takes into account the one-dimensional topology of DNA and includes molecular events known to occur in genome dynamics over the course of evolutionary time.
We here summarize the most essential types of genomic phenomena which are necessary for the emergence of fractality. Let us consider a sequence where a population of markers (representing the members of a TE population) is randomly distributed. We assume the following molecular events, each with an assigned probability of occurrence:
- (a)
Elimination of a marker (repeat) of the initial population, which occurs either by recombinational excision [7], or due to progressive decomposition by nucleotide substitutions and/or indel events. This leads to the aggregation of the spacers initially separated by the eliminated repeat.
- (b)
Incorporation into existing spacers of “Subsequently Inserted genomic Material” (SIM) such as: repeats of more recent TE families, viral or other exogenous DNA etc. Each genomic locus has the same probability of SIM incorporation, i.e., larger spacers have higher probability of SIM incorporation.
- (c)
In some simulations we further include transposition events of parts of the sequence, which are randomly cut from their original position and inserted randomly in a new position. These latter phenomena do not represent integral part of the model and are studied because they could lead to destruction of fractality due to ongoing randomization. Such events are known to happen in genomes over the course of evolutionary time and represent a naturally occurring “shuffling” of the genomic structure.
The plots, as well as the presented regression analyses were performed using Grace-5.1.14, which is a free-code plotting tool for X Windows Systems [24]. Programs in FORTRAN and C and scripts in Perl are available upon request.
3. Results
In the present study chromosomes are treated as symbol-sequences by replacing nucleotides belonging to a considered TE type with 1s while replacing with 0s the rest of the nucleotides. This is performed either in all repeat copies or excluding the most deteriorated and/or truncated ones (for details see the Methods). This treatment is suggested by the finding that, always, the most extended power-laws in inter-repeat distances’ size distributions are found excluding the most deteriorated and/or truncated copies. As we discuss later on, this property is in accordance with the model we propose for the generation of fractality and long-rangeness in genomes (see following sections and references [10,11]).
3.1. Scaling Properties of Block Entropy in TE Genomic Distributions
In Figure 1, we present examples of H(n) plotted versus n for chromosomes from different organisms. The full list of examined cases is given in Table 1. Most cases of well-shaped fractality are found in small genomes (A. thaliana. C. elegans, D. melanogaster, O. sativa). However, small genomes have relatively low populations of repeats, usually of the order of tenths per chromosome for every repeat type. Thus repeat populations suitable for our analysis are scarce in small genomes. All these cases (relatively few, see Table 1) present a well-shaped linear region and high values of R indicating an entropic scaling compatible with fractality in the chromosomal distribution of repeats. On the other hand, in large genomes (the large majority of eukaryotic ones) the occurrence of important linearity in the entropic scaling is scarce, and is relatively frequent only in the human genome, which is well known for large populations of highly inhomogeneously distributed repeats, often following clustering at several length scales [7,10].
The quantification of the results of the application of the entropic scaling in the genomic distribution of repeats is not entirely straightforward. The related literature [3–6,12–15] and our earlier results [2] applying the entropic scaling on by-construction fractal symbol sequences and on the chromosomal distribution of protein-coding segments show that linearity in semilogarithmic scale is nearly equivalent to fractality and to the existence of long-range order. Moreover, another indication that the genomic repeat distribution forms an ordered structure is provided by low entropy values of chromosomes vs. the entropy value for same word length n of the corresponding surrogate sequence presented in all of our entropy scaling plots. So, in Table 1, the results of entropic scaling are quantified by three measures: (a) The extent of linearity in semi-logarithmic scale (E); (b) The corresponding slope (S); (c) The ratio (R), as defined in the Methods, with high values of R indicating fractality.
Six out of the thirty-three examined cases present a sizable linear region on the plot (one and a half order of magnitude or higher), while another ten exhibit a smaller but clearly distinctive linear segment. All the plots corresponding to the rows of Table 1 are included in the supplementary material. In all of these plots, the entropic scaling curve for the corresponding surrogate data (artificial chromosome with randomly positioned repeats, see Methods) is included. Note that while linearity in semi-logarithmic scale does not represent a general rule (only 16 out of 33 cases), in all examined chromosomes the entropy values for the genomic sequence are clearly lower than the ones of the surrogate data (for same values of word length n). Thus, genomic sequences have strongly reduced entropies compared to their random counterparts, indicating an important degree of internal structure.
3.2. Fractality in Repeat Genomic Distributions, as Measured by a Box-Counting Method
In Figure 2 are presented the eight box-counting plots for the same chromosomes shown in Figure 1. In the supplementary material, the full collection of plots is included. In all plots, we observe two linear segments, in the low and the high value regions of the box size. The slopes for the low-length segments always are between −0.9 and −1. In the high-length region the extent and the slope of the linear segment vary considerably, and it is in this region where we expect fractality. Extent of linearity (F1 and F2) and the fractal (similarity) dimension measured by the corresponding slopes of the two linear segments (D1 and D2) are given in Table 1. In 15 out of the 33 examined eukaryotic chromosomes (from 13 organisms) the slope D2 has an absolute value (corresponding to Df) lower than 0.9. In 11 cases Df does not exceed the value 0.7, while the extent of linearity may reach or exceed three orders of magnitude. In eight cases we have an extent larger than two orders of magnitude, while the fractal dimension is 0.7 or lower, thus indicated a well-shaped fractal structure. Seven out of eight such cases are met in small genomes (A. thaliana, C. elegans, D. melanogaster, O. sativa) and the last one in a H. sapiens chromosome. In the full set of 15 cases showing fractality, again small genomes are overrepresented, while the human genome represents a maximum for the large eukaryotic genomes.
In the first ten full chromosomes included in Table 1, out of the complete set of 33 examined cases, we did the following modifications in our methodology in order to further elaborate on the distinct features of the box-counting curves:
- (a)
Shuffling (random rearrangement) of the repeat population inside the initial chromosome.
In the box-counting curves the linear segment corresponding to the high-length region acquires, invariably, a slope close to −1, as expected (fractality disappears).
- (b)
Replacement of each repeat by a single “1” symbol.
When repeats are replaced by a single “1” symbol, the box-counting curves lose their low-length linear segment.
- (c)
Replacement of each repeat by a single “1” symbol followed by shuffling (random rearrangement) of the repeat population inside the initial chromosome.
Due to the combination of both interventions (a) and (b), box-counting curves lose their low-length-region linear part and the large length linear region gets a slope close to −1.
The related quantitative information for these manipulated (surrogate) genomic sequences is given in Table 2. In Figure 3 the three plots for the chromosomal distribution of the DNA transposon CELE14B in chromosome 5 of C. elegans (row 6 in Table 2) are depicted. The full set of the plots corresponding to the manipulations above is given in the supplementary material. The inclusion in our study of the box-counting analysis based on the surrogate data sets (a), (b) and (c) leads to the conclusion that the linear segment in log-log plots, always found in the low-length region, relies on the size distribution of the repeats themselves. As this size distribution is short ranged (all repeats being more or less truncated copies of the same ancestral sequence, see e.g., [8]) it lacks any trace of fractality, as shown by the corresponding absolute slopes (see the D1 values in Table 1), which are always near-unity.
3.3. Study of Chromosomal Regions from Chromosomes without Indications of Fractality when Studied in Their Entirety
We apply the block entropy scaling and box-counting methodology to four chromosomal regions from chromosomes which, as a whole, do not present indications of fractality. The criterion for selecting the chromosomal regions was a relatively high percentage of repeat insertions which occurred after the proliferation of the examined TE population. In all four cases both methods of entropic scaling and box-counting show well developed fractality. The plots of these chromosomal regions are given in the supplementary material while one such case is presented in Figure 4.
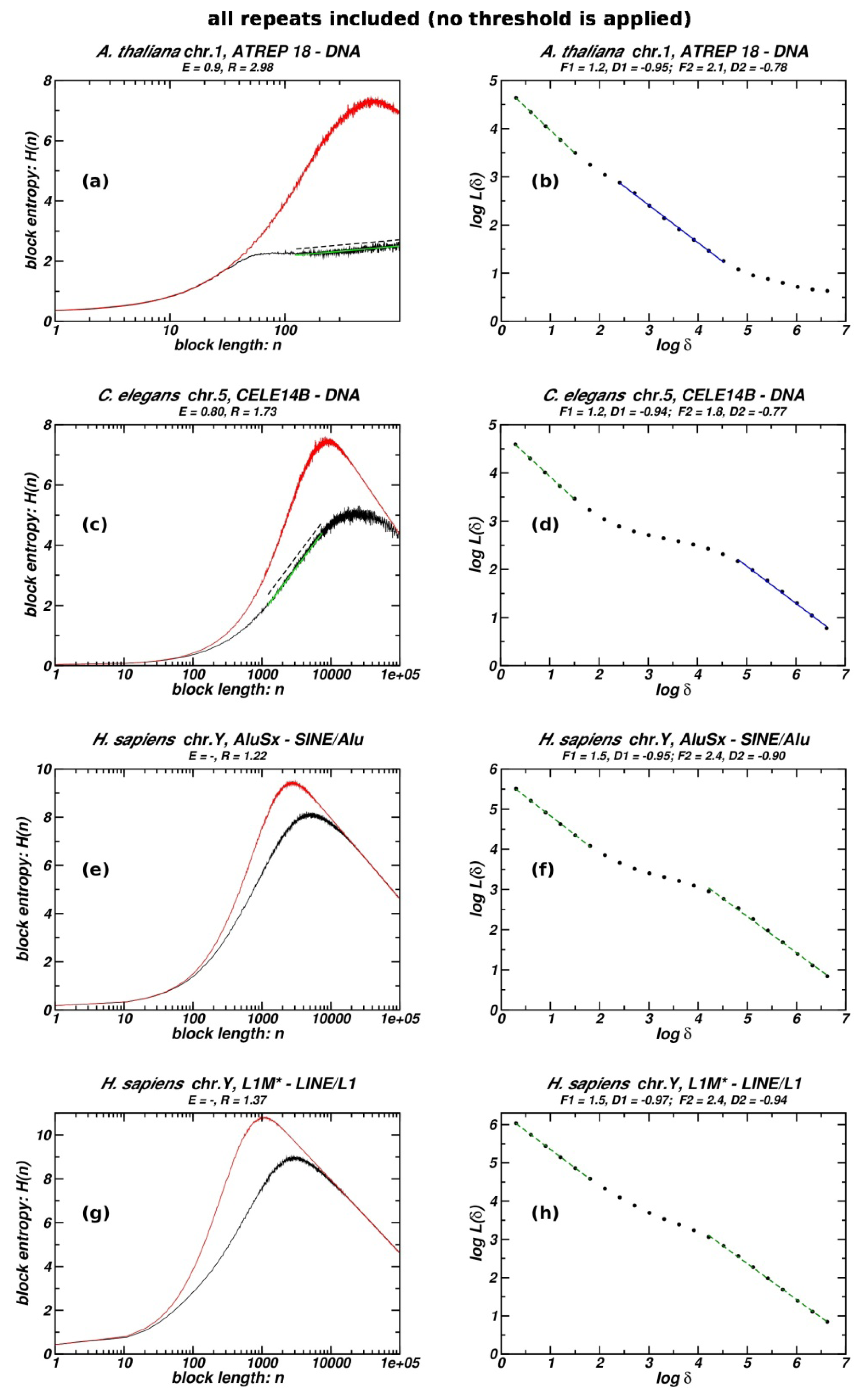
3.4. Entropic Scaling and Box-Counting for Whole TE Populations without Excluding the Heavily Deteriorated or Truncated Repeat Copies
The repeat populations (of several TE types) per chromosome studied and listed in Table 1 are the ones where heavily deteriorated or truncated repeat copies are excluded, as explained in the Methods. Preliminary tests have shown that the adoption of threshold values for Divmax or Lenmin, which are found to optimize the extent of linearity in log-log scale in the inter-repeat distance distributions [10,11], is also efficient for the study presented herein. In order to assess the impact of such thresholds in our study, we analyse the entropic scaling and box-counting in four cases from Table 1. In Figure 5 we present plots for the whole TE populations, and in Table 3 quantitative results are shown along with the corresponding quantities when thresholds excluding deteriorated and truncated copies are imposed
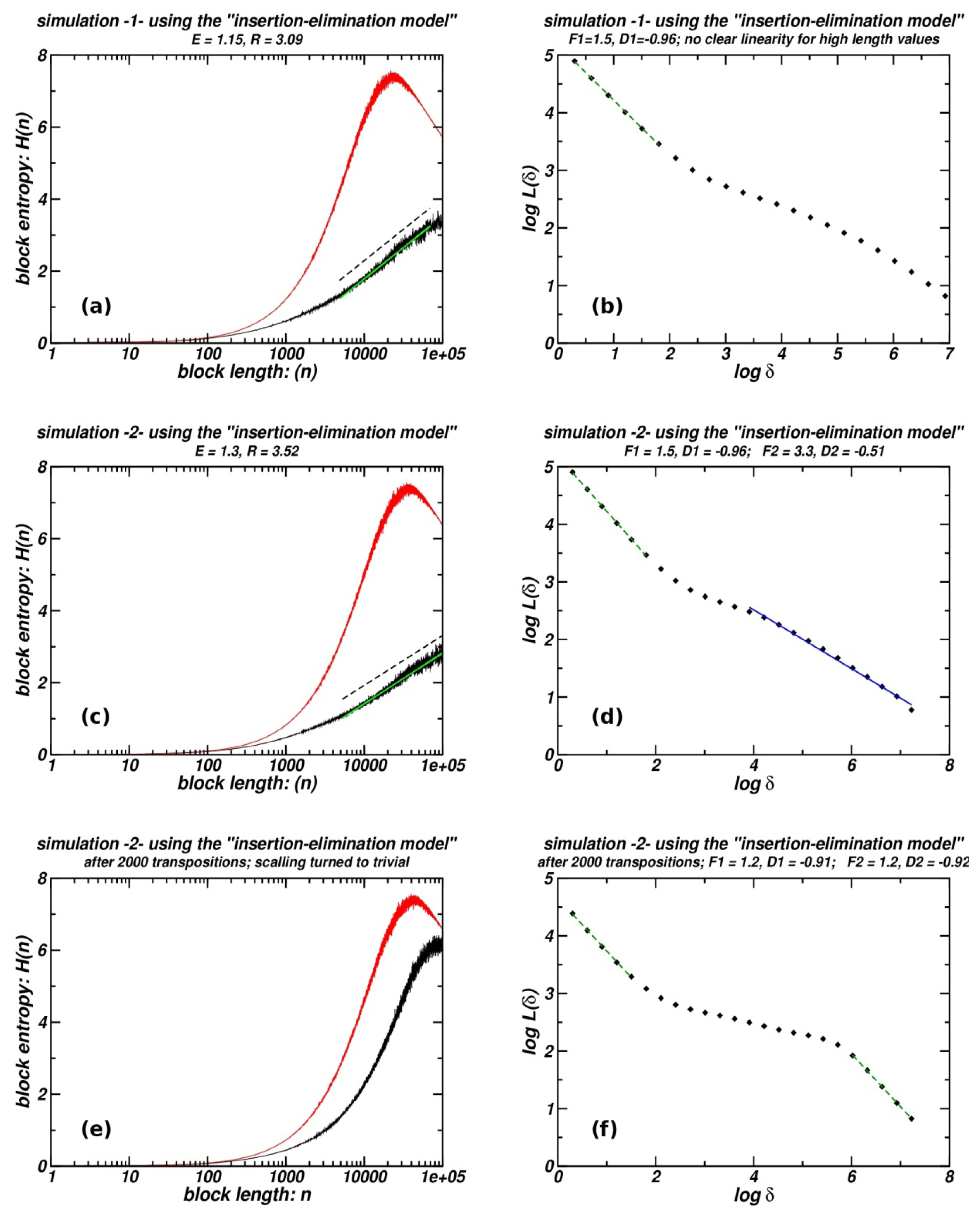
3.5. Entropic Scaling and Box-Counting for Sequences Produced Using the “Insertion-Elimination Model”
In Figure 6, results from two simulations in which the initial sequence comprises a random distribution of TEs are presented. They correspond to the first two rows of plots (a–b, c–d) where the entropic and box-counting plots of the final product of the application of the model are included. The second numerical experiments differ from the first only in the amount of the genomic material (SIM) inserted during the simulation (two-fold increase), while the initial and final number of markers are set to be equal. It is clear that increase of SIM entails more pronounced fractality (for details of the simulations see in the supplementary material). Increases of the obtained fractality may be also attained when further eliminations of repeats belonging to the initial set are allowed. Thus, a higher influx of subsequently inserted sequences, more intense elimination rates, or longer maturation in time of a repeat (TE) population, all should contribute to the formation of a more extended fractal structure. The inversion of this procedure, leading finally to the destruction of any fractal pattern is attained when a sufficient number of transposition events (genomic shuffling) are simulated, as shown in the third row (e–f). These plots correspond to a sequence obtained after 2000 random transpositions of sequence-segments, where the product of the second numerical experiment (c–d) is used as an initial “artificial chromosome”. While this amount of genomic shuffling suffices to destroy fractality as shown in plots e and f, lower numbers of transposition events result into a reduced extent of fractality before reaching its complete destruction (figures not shown). In the entropic scaling plots of Figure 6, curves corresponding to surrogate random sequence are also included.
4. Discussion
4.1. Understanding the Entropic Scaling and Fractality of TE Chromosomal Distribution—Are the Shown Results Compatible with the Proposed Model?
By calculating the block - Shannon entropy we can determine the scaling that is hidden within any kind of symbol-sequence, which reflects a complex process entailing scale invariance and fractality [25]. Thus, during the last decades, the entropic analysis has been used in time series from signal transmission in electronic engineering, in earthquakes [26,27], in economy [28] and in many other fields from physics and physiology [29] and to social sciences [30]. More specifically, in block entropy studies traditionally the entropy values are computed using a constant block (word) length comparing different segments of time series or symbol sequences in order to estimate the information content per block, see e.g., [27]. Alternatively, as we implemented in our study, entropic values are computed with variable block length windows and fractality and self-similarity are estimated by the whole time series. For another application of entropic scaling where a long memory has been observed in time series analysis see [30] in the context of a sociology study. Box-counting is a standard technique that has been used to find and measure fractality in patterns that show scale-free geometries such as geographical landscapes materials, surfaces and biological systems like the lung and the blood circulatory system [16,17,31].
In the presented results, we see that linearity in semi-logarithmic scale in entropic scaling plots and linearity in double-logarithmic scale in box-counting plots with the absolute slope considerably diverging from unity are present in several cases of complete chromosomes (see Table 1 and Figures 1 and 2). This observation raises several questions, such as about the causes of the differences found between genomes and about the causes of the systematically higher incidence of fractality in small genomes (and consequently in small chromosomes). Additionally, in cases where fractality is observed, the intensity and resolution of this pattern (as measured by E and R) is unevenly distributed between different genomes. Another result of the present study is the relative scarcity of fractality studied herein compared to observations of long-ranged order in studies at the whole-chromosome level of inter-repeat distances [10,11]. Occurrence of power-law-like distributions is much more frequent than fractality. Another interesting observation is that often, although a fractal pattern is observed in regions of large chromosomes, it is not seen when the whole chromosome is considered. In the present section we try to investigate the complete picture that emerges when we combine the information gathered from all the setups examined herein and to assess the ability of the proposed “insertion-elimination model” to reproduce the observed fractal or fractal-like patterns.
Recapitulating the essence of the proposed model, the necessary and sufficient conditions for the emergence of box-counting fractality and for the observed entropic scaling are the concurrent occurrence of (a) repeat eliminations and (b) inflation of the sequence, at least for some periods of the genomic evolution. Sequence length increase may be due to subsequent insertions of repeats belonging to more recent TE families or other molecular events, such as: viral sequences incorporated into the host genome, segmental duplications [32,33], sporadic occurrence of whole genome duplications [34,35] and local growth of the sequence due to microsatellite proliferation [36].
Fractality in a chromosomal region combined with absence of fractality in the whole chromosome is shown in Figure 4 and in three other similar cases presented in the supplementary material. This finding advocates in favor of the importance of high insertion rates of more recent TE families for the formation of a well-shaped fractal structure in the distribution of older TE families. Moreover, other sources of sequence insertion may also have contributed toward this direction. Another aspect of this finding is revealed if we take into account that, while the entire chromosome in all four cases lacks any trace of fractality, when studied from the point of view of the existence and extent of a power-law in the inter-repeat spacers sizes, the difference between chromosomal regions and whole chromosomes is milder, although pointing towards the same direction [11]. i.e., in all four cases, the extent of linearity in double-log scale is larger in the insertion-rich regions than in the entirety of the chromosome, but complete chromosomes are still characterized by power-laws in the inter-repeat size distributions. The absence of fractality in these four complete chromosomes and the reported overall scarceness of the fractal pattern in large chromosomes has to be contrasted with the more frequent occurrence of fractal geometry in small genomes. These observations lead us to the conjecture that large chromosomal size drastically undermines fractality, but does not hinder the development of power-laws in the inter-repeat size distributions. Note that the size of chromosomes in small genomes (e.g., Caenorhabditis elegans and Arabidopsis thaliana) is of the same order of magnitude as the chromosomal regions studied herein (a few tenths of millions of nucleotides), while entire chromosomes of large (e.g., mammalian) genomes are larger by one order of magnitude. We have to emphasize however, that chromosomes of small genomes are not equivalent with chromosomal regions of large genomes. Large genomes are well known to be subjected to intense shuffling during their evolution due to frequent intra- and inter-chromosomal rearrangements and translocations. Thus, they might preserve fractality in chromosomal regions while tend to destroy the whole-chromosome fractal pattern. On the other hand, in species with small genomes and high effective population sizes, purifying selection prevents extensive intra- and inter-chromosomal rearrangements, thus preserving an emerging fractal-like pattern. Moreover, a high rate of repeat loss driven by natural selection has been reported for the D. melanogaster genome [37] and a similar trend is observed in other genomes too, which have been subjected to size reduction recently. These characteristics of small genomes in combination with frequent incidence of fractality therein corroborate the proposed model. Note that the appearance of power-laws in inter-repeat spacers’ size distributions is expected to be less affected by rearrangements: each rearrangement event may affect the size of only one inter-repeat spacer, while it seriously perturbs the whole fractal pattern estimated by means of box-counting.
In Figure 5 one can see the entropic scaling and the box-counting plots for four repeat populations, where, unlike cases included in Table 1 and Figures 1 and 2, no thresholds of maximum divergence or minimum length are applied. Here, whole repeat (TE) populations of each chromosome are studied, not excluding the more truncated or deteriorated copies. Data for the quantitative comparison of whole and threshold-limited cases are provided in Table 3. Fractality is quantified by the box-counting slope D2 and the corresponding linearity F2; and by the extent of the linear segment E and the ratio R in entropic scaling plots. In cases of DNA Transposable Elements (lines 1, 2 in Table 3) filtering of deteriorated TE copies reinforces fractality, while the whole repeat populations still exhibit fractality. When we examine retroelements, like case 3 (AluSx belonging to the Short INterspersed Elements, SINEs) and case 4 (L1M belonging to the Long INterspersed Elements, LINEs) in Table 3, the contrast is higher: the filtered repeat populations exhibit well-shaped fractality as assessed by the above criteria, while whole chromosomal populations fail to form any fractal pattern. The conclusion drawn above is corroborated by several other cases of comparison between whole and threshold-limited TE populations not included herein. The observed difference between whole and filtered TE populations is compatible with the hypothesis underling the proposed model; i.e., that the formation of the fractal structure depends positively on the elimination rate of the studied TE population. The reason for the dependence and the impact of the different modes of repeat eliminations on the different classes of TEs in view of the “insertion-elimination model” are detailed in [11], while they may briefly explained here as follows: one important mechanism in the elimination of repeats over the course of evolutionary time is the recombinational excisions of members of the same TE population when they are located close to one another at opposite orientations (inverted pairs). Such molecular events are facilitated by close resemblance in the primary structure (nucleotide sequence) of the “mutually annihilated” TE copies [7,38]. Several other recombinational interactions between TE copies of the same population contributing to repeat eliminations always rely on sequence integrity and similarity between the repeats interacting through recombination. Differences between DNA-TEs and retroelements in their response towards the inclusion in the studied population of truncated and deteriorated copies can be explained on the basis of the high dependence of the retroelements’ eliminations of recombination events and the clearly lower incidence of recombination driven eliminations of DNA TEs, see the supplementary file 4 Section 6 “DNA transposons in mammalian genomes” in [11]. For a more detailed study of the dynamics of DNA TEs in the human genome, the reader may refer to page 880 of reference [36]. As a consequence of these differences, the corresponding disparity in the propensity for the formation of fractal structures as shown in Table 3 between whole and filtered TE populations is compatible with the insertion-elimination model proposed herein.
4.2. The “Fractal Globule” Model for the Eukaryotic Nucleus—Possible Role of the Chromosomal Distribution of Transposable Elements
The fractal globule model for the folding of chromatin in the eukaryotic nucleus predicts a power-law distribution of the genomic distance and contact probability which has been observed experimentally [39]. A fractal structure of the genome has been theoretically predicted a long time ago [40,41] and seems to offer important benefits to cellular functioning. Such a knot-free structure makes possible the repetitive winding and unwinding of the genome during consecutive cell-cycles [42]. The predicted scaling features of the fractal globule have been quantitatively verified by a combination of molecular biology and computational techniques [39,43]. Recently, the 3D clustering of repeats belonging to the same family due to repeat pair interactions has led to the suggestions that such repeats coordinate and maintain the chromatin higher structure [44]. Presumably, fractality and long-rangeness initially developed through neutral genome dynamics such as the ones related to repeat proliferation we discussed, and subsequently were preserved by selective forces, as this structure is intertwined with multiple genomic functions. The long-ranged distribution of populations of highly similar sequence segments, such as TEs, could help the initial shaping and maintenance of the fractal globule state, by means of recombinational DNA-DNA “kissing interactions” [45]. Ancient TE populations could have participated in the fractal globule formation, while subsequent repeat families might continue to contribute to its reshaping. We have seen that there is no universal exponent (slope in double logarithmic scale) across all species and TEs classes studied herein, this finding indicating that the observed exponents reflect the combined result of both, adaptive and neutral molecular dynamic processes.
5. Conclusions
We conducted a study of the genomic distribution of transposable element in whole chromosomes using entropic scaling analysis and box-counting. Repeat populations from all the main TE classes are considered within several genomes from taxonomically distant organisms, and employ two different methodologies, box-counting and entropic scaling. Box-counting is a standard tool for the identification of fractality and for its quantification through values of the absolute value of the slope (fractal dimension) and extent of linearity in double logarithmic scale [16,17]. Entropic scaling as described in the cited literature (see e.g., [3,4,14,15]) also represents an indication of the emergence of self-similarity and is tightly correlated with fractality.
This study expands findings of previous works of our group where the positioning of TEs in whole genomic sequences was analysed by studying the distributional patters of the lengths of inter-repeat distances [10,11]. There, power-law-like distributions were often observed. However, power-law-like distributions, linear entropic scaling curves in semi-logarithmic scale and linearity in log-log scale with a fractal dimension substantially lower than unity are not equivalent, but form a nested hierarchy with the power-law-like pattern in the inter-repeat distances being relatively more frequent and fractality derived using box-counting being the more scarce. We find a connection between the frequency of fully developed fractality and the genome size, smaller genomes being the more prone to develop fractality along with some of the large ones, like the human genome. Also, we suggest that fractality in the repeat distribution might have contributed to the shaping of the recently found fractal globule geometry of the chromatin folding within the eukaryotic nucleus.
Acknowledgements
The authors would like to thank Jaffar Hasnain and Dimitris Polychronopoulos for useful insights and fruitful discussions. Part of this work was performed in the framework of “Target Identification for Disease Diagnosis and Treatment (DIAS)” project within GSRT’s KRIPIS action, funded by Greece and the European Regional Development Fund of the European Union under the O.P. Competitiveness and Entrepreneurship, NSRF 2007–2013.
Conflicts of Interest
The authors declare no conflict of interest.
- Author ContributionsAll authors have equally contributed to the to conception and design of this work, acquisition of data and analysis and interpretation of data.
- Supplementary MaterialsSupplementary materials can be accessed at: https://www.mdpi.com/1099-4300/16/4/1860/s1.
References
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J 1948, 27, 379–423. [Google Scholar]
- Athanasopoulou, L.; Athanasopoulos, S.; Karamanos, K.; Almirantis, Y. Scaling properties and fractality in the distribution of coding segments in eukaryotic genomes revealed through a block entropy approach. Phys. Rev. E 2010, 82, 051917. [Google Scholar]
- Grassberger, P. Toward a quantitative theory of self-generated complexity. Int. J. Theor. Phys 1986, 25, 907–938. [Google Scholar]
- Ebeling, W.; Nicolis, G. Entropy of symbolic sequences: The role of correlations. Europhys. Lett 1991, 14, 191–196. [Google Scholar]
- Ebeling, W.; Nicolis, G. Word frequency and entropy of symbolic sequences: A dynamical perspective. Chaos Solitons Fractals 1992, 2, 635–650. [Google Scholar]
- Ebeling, W.; Freund, J.A.; Rateitschak, K. Entropy and extended memory in discrete chaotic dynamics. Int. J. Bifurcat. Chaos 1996, 6, 611–625. [Google Scholar]
- Jurka, J.; Kohany, O.; Pavlicek, A.; Kapitonov, V.V.; Jurka, M.V. Duplication, coclustering, and selection of human Alu retrotransposons. Proc. Natl. Acad. Sci. USA 2004, 101, 1268–1272. [Google Scholar]
- Jurka, J.; Kapitonov, V.V.; Kohany, O.; Jurka, M.V. Repetitive sequences in complex genomes: Structure and evolution. Annu. Rev. Genomics Hum. Genet 2007, 8, 241–259. [Google Scholar]
- Deininger, P.L.; Batzer, M.A. Mammalian retroelements. Genome Res 2002, 12, 1455–1465. [Google Scholar]
- Sellis, D.; Provata, A.; Almirantis, Y. Alu and LINE1 distributions in the human chromosomes: Evidence of global genomic organization expressed in the form of power laws. Mol. Biol. Evol 2007, 24, 2385–2399. [Google Scholar]
- Klimopoulos, A.; Sellis, D.; Almirantis, Y. Widespread occurrence of power-law distributions in inter-repeat distances shaped by genome dynamics. Gene 2012, 499, 88–98. [Google Scholar]
- Karamanos, K.; Nicolis, G. Symbolic dynamics and entropy analysis of Feigenbaum limit sets. Chaos Solitons Fractals 1999, 10, 1135–1150. [Google Scholar]
- Ebeling, W. Entropies and Predictability of Nonlinear Processes and Time Series. In ICCS 2002, LNCS 2331; Sloot, P.M.A., Tan, C.J.K., Dongarra, J.J., Hoekstra, A.G., Eds.; Springer-Verlag: Berlin, Germany, 2002; pp. 1209–1217. [Google Scholar]
- Ebeling, W.; Rateitschak, K. Symbolic dynamics, entropy and complexity of the Feigenbaum map at the accumulation point. Discret. Dyn. Nat. Soc 1998, 2, 187–194. [Google Scholar]
- Nicolis, G.; Gaspard, P. Toward a probabilistic approach to complex systems. Chaos Solitons Fractals 1994, 4, 41–57. [Google Scholar]
- Mandelbrot, B.B. The Fractal Geometry of Nature; W.H. Freeman: San Francisco, CA, USA, 1982. [Google Scholar]
- Feder, J. Fractals; Plenum Press: New York, NY, USA, 1988. [Google Scholar]
- University of California Santa Cruz (UCSC) Genome Browser. Available online: http://www.genome.ucsc.edu accessed on 1 September 2010.
- National Center for Biotechnology Information. Available online: ftp://ftp.ncbi.nih.gov/genomes accessed on 1 November 2010.
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0, Available online: http://www.repeatmasker.org accessed on 1/11/2010.
- Jurka, J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet 2000, 16, 418–420. [Google Scholar]
- Gish, W. WU-BLAST 20, 2003. Available online: http://blast.wustl.edu accessed on 1 November 2010.
- Takayasu, H.; Takayasu, M.; Provata, A.; Huber, G. Statistical properties of aggregation with injection. J. Stat. Phys 1991, 65, 725–745. [Google Scholar]
- Grace, Available online: http://plasma-gate.weizmann.ac.il/Grace accessed on 1 March 2007.
- Scafetta, N. An Entropic Approach to the Analysis of Time Series. In Ph.D. Thesis; University of North Texas: Denton, TX, USA, 2001. [Google Scholar]
- Karamanos, K.; Peratzakis, A.; Kapiris, P.; Nikolopoulos, S.; Kopanas, J.; Eftaxias, K. Extracting preseismic electromagnetic signatures in terms of symbolic dynamics. Nonlinear Process. Geophys 2005, 12, 835–848. [Google Scholar]
- Bezerianos, A.; Tong, S.; Thakor, N. Time-dependent entropy estimation of EEG rhythm changes following brain ischemia. Ann. Biomed. Eng 2003, 31, 221–232. [Google Scholar]
- Marschinski, R.; Kantz, H. Analysing the information flow between financial time series: An improved estimator for transfer entropy. Eur. Phys. J. B 2002, 30, 275–281. [Google Scholar]
- Kurths, J.; Voss, A.; Witt, A.; Saparin, P.; Kleiner, H.J.; Wessel, N. Quantitative analysis of heart rate variability. Chaos 1995, 5, 88–95. [Google Scholar]
- Scafetta, N.; Hamilton, P.; Grigolini, P. The thermodynamics of social processes: The teen birth phenomenon. Fractals 2001, 9, 193–208. [Google Scholar]
- Havlin, S.; Buldyrev, S.V.; Goldberger, A.L.; Mangegna, R.N.; Ossadnik, S.M.; Peng, C.-K.; Simons, M.; Stanley, H.E. Fractals in biology and medicine. Chaos Solitons Fractals 1995, 6, 171–201. [Google Scholar]
- McLysaght, A.; Hokamp, K.; Wolfe, K.H. Extensive genomic duplication during early chordate evolution. Nat. Genet 2002, 31, 200–204. [Google Scholar]
- De Grassi, A.; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene families. Gene 2008, 421, 1–6. [Google Scholar]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant. Biol 2005, 8, 135–141. [Google Scholar]
- Sémon, M.; Wolfe, K.H. Reciprocal gene loss between Tetraodon and zebrafish after whole genome duplication in their ancestor. Trends Genet 2007, 23, 108–112. [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921.
- Petrov, D.A.; Aminetzach, Y.T.; Davis, J.C.; Bensasson, D.; Hirsh, A.E. Size matters: Non-LTR retrotransposable elements and ectopic recombination in Drosophila. Mol. Biol. Evol 2003, 20, 880–892. [Google Scholar]
- Lobachev, K.S.; Stenger, J.E.; Kozyreva, O.G.; Jurka, J.; Gordenin, D.A.; Resnick, M.A. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J 2000, 19, 3822–3830. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- Grosberg, A.; Nechaev, S.K.; Shakhnovich, E.I. The role of topological constraints in the kinetics of collapse of macromolecules. J. Phys. France 1988, 49, 2095–2100. [Google Scholar]
- Grosberg, A.; Rabin, Y.; Havlin, S.; Neer, A. Crumpled globule model of the three-dimensional structure of DNA. Europhys. Lett 1993, 23, 373–378. [Google Scholar]
- Vasilyev, O.A.; Nechaev, S.K. On topological correlations in trivial knots: New arguments in support of the crumpled globule concept. Theor. Math. Phys 2003, 134, 142–159. [Google Scholar]
- Mateos-Langerak, J.; Bohn, M.; de Leeuw, W.; Giromus, O.; Manders, E.M.; Verschure, P.J.; Indemans, M.H.; Gierman, H.J.; Heermann, D.W.; van Driel, R.; Goetze, S. Spatially confined folding of chromatin in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 3812–3817. [Google Scholar]
- Tang, S.-J. Chromatin Organization by Repetitive Elements (CORE). A genomic principle for the higher-order structure of chromosomes. Genes 2011, 2, 502–515. [Google Scholar]
- Kleckner, N.; Weiner, B.M. Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic, and premeiotic cells. Cold Spring Harb. Symp. Quant. Biol 1993, 58, 553–565. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Organism | Chromosome | Repeat Name | Family Name | Divmax or Lenmin | E | S | R | F1 | D1 | F2 | D2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ath | Chr01 | ATREP18 | DNA | Divmax = 21% | 1,00 | 0,26 | 2,58 | 1,2 | −0,95 | 3,7 | −0,65 |
| 2 | Ath | Chr01 | COLAR12 | Satellite/Centr | Divmax = 25% | 2,73 | 0,27 | 3,9 | 1,2 | −0,96 | 3,6 | −0,68 |
| 3 | Bta | Chr12 | L2c | LINE/L2 | Lenmin = 0 | 0 | 1,14 | 1,2 | −0,94 | 2,1 | −0,94 | |
| 4 | Bta | Chr25 | Bov-tA2 | SINE/BovA | Divmax = 20% | 0 | 1,09 | 1,2 | −0,95 | 3 | −0,95 | |
| 5 | Cel | Chr01 | CERP3 | Unknown | Divmax = 26% | 0,87 | 1,22 | 1,7 | 1,5 | −0,94 | 2,1 | −0,7 |
| 6 | Cel | Chr05 | CELE14B | DNA | Divmax = 8% | 1,37 | 0,701 | 2,08 | 1,5 | −0,91 | 2 | −0,6 |
| 7 | Cfa | Chr01 | MER20 | DNA/MER1_type | Divmax = 26% | 0 | 1,18 | 1,2 | −0,94 | 1,5 | −0,92 | |
| 8 | Cfa | Chr01 | L3 | LINE/CR1 | Lenmin = 0 | 0 | 1,15 | 1,2 | −0,95 | 2,1 | −0,92 | |
| 9 | Cfa | Chr20 | L1_Canis1 | LINE/L1 | Lenmin = 600 | 0,85 | 1,46 | 1,53 | 2,1 | −0,98 | 0 | - |
| 10 | Cfa | ChrX | L1MC4 | LINE/L1 | Lenmin = 200 | 0 | 1,31 | 1,8 | −0,95 | 1,8 | −0,85 | |
| 11 | Dme | Chr2L | DNAREP1_DM | Helitron | Lenmin = 300 | 2,1 | 0,498 | 3,34 | 1,5 | −0,97 | 2,7 | −0,51 |
| 12 | Dme | Chr3R | DNAREP1_DM | Helitron | Lenmin = 200 | 1,46 | 0,588 | 3,07 | 1,5 | −0,95 | 3 | −0,35 |
| 13 | Dre | Chr01 | TC1DR3 | DNA/Tc1 | Divmax = 11% | 0 | 1,19 | 1,5 | −0,97 | 1,8 | −0,92 | |
| 14 | Gga | Chr08 | Mariner1b_GG | DNA/Mariner | Lenmin = 50 | 0,93 | 1,39 | 1,39 | 1,8 | −0,94 | 1,8 | −0,7 |
| 15 | Gga | Chr13 | Mariner1_GG | DNA/Mariner | Lenmin = 200 | 1,25 | 0,709 | 1,8 | 1,5 | −0,97 | 0 | - |
| 16 | Mdo | Chr06 | MAR1a_Mdo | SINE/MIR | Divmax = 18% | 0 | 1,2 | 1,2 | −0,95 | 3 | −0,94 | |
| 17 | Mdo | ChrX | L3_Mars | LINE/CR1 | Lenmin = 100 | 0 | 1,22 | 1,5 | −0,96 | 2,1 | −0,95 | |
| 18 | Mmu | Chr09 | B4A | SINE/B4 | Divmax = 30% | 0 | 1,27 | 1,2 | −0,95 | 2,4 | −0,93 | |
| 19 | Mmu | Chr12 | B3 | SINE/B2 | Divmax = 25% | 0 | 1,34 | 1,2 | −0,94 | 2,4 | −0,92 | |
| 20 | Osa | Chr01 | RIRE3A_LTR | LTR/Gypsy | Divmax = 7% | 0,8 | 0,732 | 2,47 | 2,1 | −0,98 | 3,3 | −0,31 |
| 21 | Osa | Chr11 | SEVERIN-2 | DNA | Lenmin = 100 | 0,95 | 1,74 | 1,41 | 1,5 | −0,92 | 1,2 | −0,86 |
| 22 | Ptr | Chr03 | L1P* | LINE/L1 | Lmin = 800 | 0 | 1,25 | 2,1 | −0,98 | 2,1 | −0,96 | |
| 23 | Ptr | Chr14 | AluJo | SINE/Alu | Divmax = 17% | 0 | 1,49 | 1,2 | −0,96 | 2,1 | −0,9 | |
| 24 | Rno | Chr01 | B3 | SINE/B2 | Divmax = 23% | 0 | 1,43 | 1,2 | −0,94 | 2,7 | −0,92 | |
| 25 | Rno | Chr01 | L2 | LINE/L2 | Lenmin = 50 | 0 | 1,29 | 1,2 | −0,94 | 2,7 | −0,92 | |
| 26 | Hsa | Chr19 | AluJb | SINE/Alu | Divmax = 20% | 0 | 1,17 | 1,2 | −0,96 | 2,4 | −0,95 | |
| 27 | Hsa | Chr09 | AluJo | SINE/Alu | Divmax = 15% | 0,62 | 1,66 | 1,53 | 1,5 | −0,94 | 1,8 | −0,81 |
| 28 | Has | ChrY | AluSx | SINE/Alu | Divmax = 10% | 1,96 | 0,419 | 3,39 | 1,5 | −0,95 | 2,7 | −0,52 |
| 29 | Hsa | Chr05 | AluSx | SINE/Alu | Divmax = 9% | 0 | 1,32 | 1,5 | −0,95 | 1,8 | −0,84 | |
| 30 | Hsa | Chr07 | L1P* | LINE/L1 | Lenmin = 1000 | 0 | 1,28 | 2,1 | −0,99 | 2,4 | −0,91 | |
| 31 | Hsa | Chr17 | L1P* | LINE/L1 | Lenmin = 700 | 1,2 | 1,27 | 1,61 | 2,1 | −0,98 | 1,2 | −0,9 |
| 32 | Hsa | ChrY | L1M* | LINE/L1 | Lenmin = 1300 | 1,6 | 0,854 | 1,96 | 2,1 | −0,98 | 2,1 | −0,62 |
| 33 | Hsa | Chr22 | L1M* | LINE/L1 | Lenmin = 1400 | 1,3 | 1,1 | 1,53 | 2,1 | −0,98 | 1,5 | −0,48 |
| Organism | Chromosome | Repeat Name | Family Name | Divergence or Length Threshold | F1-r [randomly rearranged repeats, extent of the 1st linear segment] | D1-r [randomly rearranged repeats, slope of the 1st linear segment] | F2-r [randomly rearranged repeats, extent of the 2nd linear segment] | D2-r [randomly rearranged repeats, slope of the 2nd linear segment] | F-1sp [each repeat replaced by a single point, extent of the linear segment] | D-1sp [each repeat replaced by a single point, slope of the linear segment] | F-rsp [randomly distributed single points, extent of the linear segment] | D-rsp [randomly distributed single points, slope of the linear segment] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ath | Chr01 | ATREP18 | DNA | Divmax = 21% | 1,2 | −0,95 | 1,2 | −0,93 | 2,1 | −0,67 | 1,2 | −0,93 |
| Ath | Chr01 | COLAR12 | Satellite/Centr | Divmax = 25% | 1,5 | −0,94 | 1,5 | −0,93 | 3 | −0,6 | 1,5 | −0,93 |
| Bta | Chr12 | L2c | LINE/L2 | Lenmin=0 | 1,2 | −0,94 | 2,1 | −0,96 | 2,1 | −0,94 | 2,1 | −0,96 |
| Bta | Chr25 | Bov-tA2 | SINE/BovA | Divmax=20% | 1,2 | −0,95 | 2,4 | −0,97 | 3 | −0,94 | 2,7 | 0,97 |
| Cel | Chr01 | CERP3 | Unknown | Divmax = 26% | 1,2 | −0,96 | 1,5 | −0,92 | 1,8 | −0,74 | 1,5 | −0,93 |
| Cel | Chr05 | CELE14B | DNA | Divmax = 8% | 1,2 | −0,94 | 0,9 | −0,92 | 2,1 | −0,6 | 1,2 | −0,9 |
| Cfa | Chr01 | MER20 | DNA/MER1_type | Divmax = 26% | 1,2 | −0,95 | 1,5 | −0,95 | 1,5 | −0,92 | 1,5 | −0,95 |
| Cfa | Chr01 | L3 | LINE/CR1 | Lenmin = 0 | 1,2 | −0,95 | 1,8 | −0,97 | 1,8 | −0,95 | 1,8 | −0,97 |
| Cfa | Chr20 | L1_Canis1 | LINE/L1 | Lenmin = 600 | 1,8 | −0,99 | 1,2 | −0,94 | 0 | 0 | 1,2 | −0,94 |
| Cfa | ChrX | L1MC4 | LINE/L1 | Lenmin = 200 | 1,5 | −0,97 | 1,5 | −0,95 | 1,8 | −0,85 | 1,5 | −0,95 |
| Cace No | Organi, Chr/me | Repeat & Family Name | Divmax or Lenmin | Tresholds (Dmax or Lmin) applied (cf. Figures 1&2) A | Whole repeat populations studied (cf. Figure 5) B | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E | R | F2 | D2 | E | R | F2 | D2 | ||||
| 1 | Ath, Chr01 | ATREP18, DNA | Divmax = 21% | 1.00 | 2.,58 | 3.7 | −0.65 | 0.9 | 2.98 | 2.1 | −0,78 |
| 2 | Cel, Chr05 | CELE14B, DNA | Divmax = 8% | 1.37 | 2.08 | 2 | −0.6 | 0.80 | 1.73 | 1.8 | −0,77 |
| 3 | Hsa, ChrY | AluSx, SINE/Alu | Divmax = 10% | 1.96 | 3.,39 | 2.7 | −0.52 | - | 1.22 | 2.4 | −0,90 |
| 4 | Hsa, ChrY | L1M, LINE/L1 | Lenmin = 1300 | 1.,6 | 1.96 | 2.1 | −0.62 | - | 1.37 | 2.4 | −0,94 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Athanasopoulou, L.; Sellis, D.; Almirantis, Y. A Study of Fractality and Long-Range Order in the Distribution of Transposable Elements in Eukaryotic Genomes Using the Scaling Properties of Block Entropy and Box-Counting. Entropy 2014, 16, 1860-1882. https://doi.org/10.3390/e16041860
Athanasopoulou L, Sellis D, Almirantis Y. A Study of Fractality and Long-Range Order in the Distribution of Transposable Elements in Eukaryotic Genomes Using the Scaling Properties of Block Entropy and Box-Counting. Entropy. 2014; 16(4):1860-1882. https://doi.org/10.3390/e16041860
Chicago/Turabian StyleAthanasopoulou, Labrini, Diamantis Sellis, and Yannis Almirantis. 2014. "A Study of Fractality and Long-Range Order in the Distribution of Transposable Elements in Eukaryotic Genomes Using the Scaling Properties of Block Entropy and Box-Counting" Entropy 16, no. 4: 1860-1882. https://doi.org/10.3390/e16041860
APA StyleAthanasopoulou, L., Sellis, D., & Almirantis, Y. (2014). A Study of Fractality and Long-Range Order in the Distribution of Transposable Elements in Eukaryotic Genomes Using the Scaling Properties of Block Entropy and Box-Counting. Entropy, 16(4), 1860-1882. https://doi.org/10.3390/e16041860