On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
“… if a closed system is at some instant in a non-equilibrium macroscopic state, the most probable consequence at later instants is a steady increase in the entropy of the system. This is the law of increase of entropy or second law of thermodynamics, discovered by R. Clausius (1865); its statistical explanation was given by L. Boltzmann in the 1870s.”
- SP1 S exists as a unique quantity for all states of the system at each instant t; consequently, it does not depend on the state of the medium;
- SP2 S is extensive, continuous and at least twice differentiable;
- SP3 S is additive over quasi-independent subsystems;
- SP4 S satisfies the second law; and
- SP5 S is nonnegative.
2. Brief Review
2.1. Equilibrium Thermodynamic Entropy
2.2. Equilibrium Statistical Entropies
3. The Second Law and Nonequilibrium States
3.1. The Second Law, non-State Entropy and Internal Variables
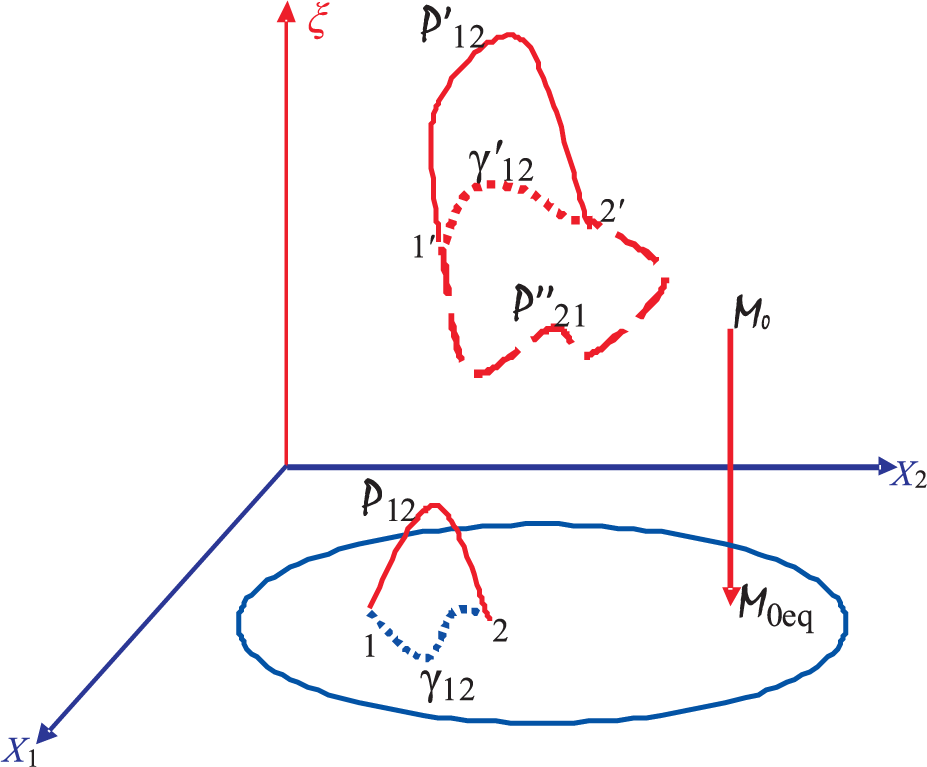
3.2. Number of Required Internal Variables
3.3. Nonequilibrium Processes
3.4. Concept of a Nonequilibrium State (NEQS) and of an Internal Equilibrium State (IEQS)
3.5. Arbitrary States as Incompletely Specified States
3.6. Internal Equilibrium versus Equilibrium
3.7. Internal Equilibrium Concept is Commonly Used
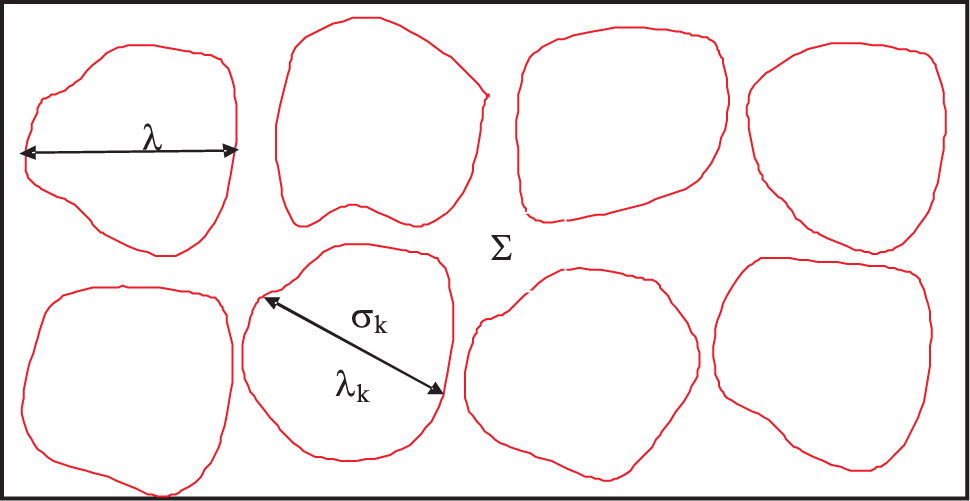
3.8. Inhomogeneous Body and Quasi-Independence
3.9. NEQS Thermodynamic Potential for an Interacting System Σ
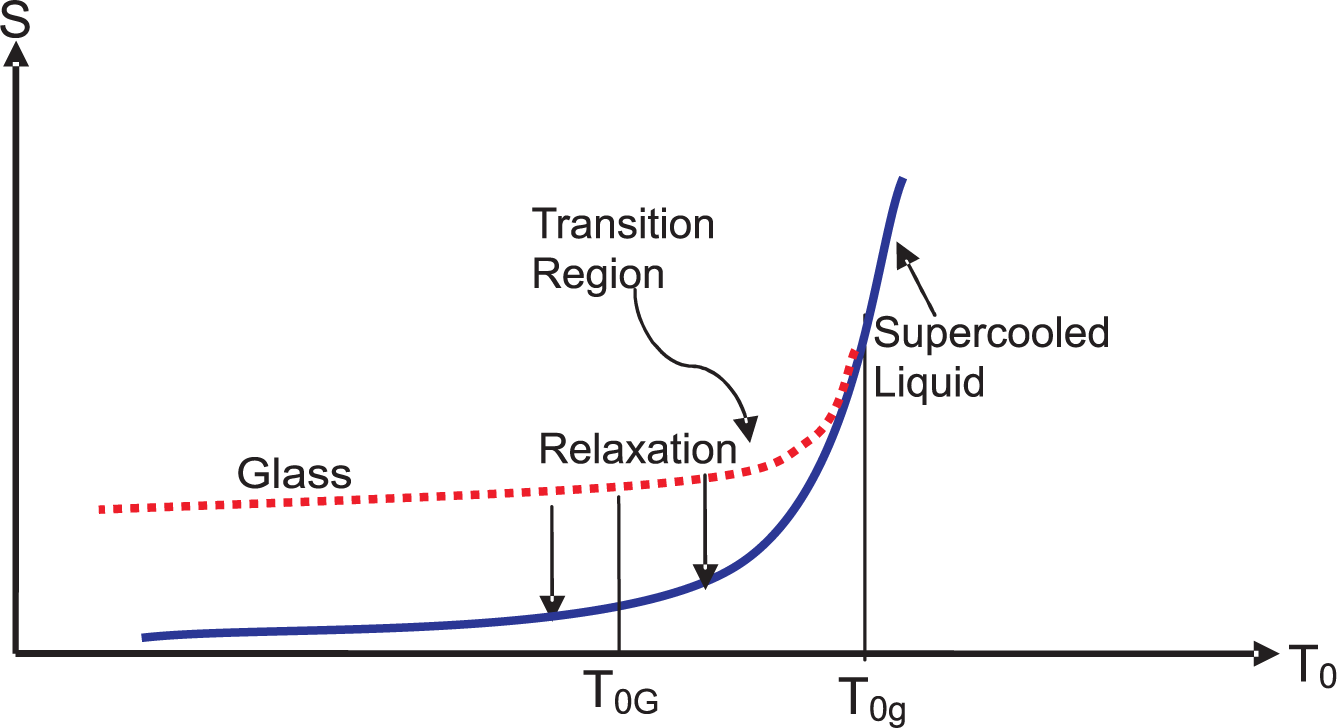
4. Vitrification and the Residual Entropy
5. Statistical Entropy for an Isolated System with a Single Component Sample Space
5.1. General Derivation of the Statistical Entropy
6. Statistical Entropy for an Interacting System with a Single Component Sample Space
6.1. Statistical Entropies of the System and Medium
6.2. Statistical Entropy as a State Function: IEQS
6.3. Statistical Entropy not a State Function: ANEQS
7. Statistical Entropy with Component Confinement
8. Semi-Classical Approximation in Phase Space for S
9. 1-d Tonks Gas: A simple Continuum Model
10. Summary and Discussion
- We need to formulate the concept of a state, state variables and state functions. We discuss these issues and the behavior of nonequilibrium thermodynamic entropy in Section 3, where we follow the recent approach initiated by us [35–39]. In particular, we discuss the role of internal variables that are needed to specify the (macro)state of a NEQS. Without internal variables, we cannot introduce the concept of a macrostate for an isolated system (see Section 3.1 and an interacting system (see Section 3.4). Thus, state variables include extensive observables (see footnote 11) and internal variables (see footnote 17) to specify a macrostate. The thermodynamic entropy is a state function for IEQS but a non-state function for ANEQS. The concept of internal equilibrium to characterize IEQSs [35–39] is based on the fact that state variables uniquely specify homogeneous macrostates. We point out its close similarity with the concept of equilibrium; see Section 3.6. The relationship of ANEQSs to IEQSs is similar to that of NEQSs to EQSs. In particular, we show how the entropy can be calculated for any arbitrary nonequilibrium process whose end states are IEQSs. We discuss various aspects of internal equilibrium and show how the entropy of an inhomogeneous system in ANEQS can be determined provided each of its subsystems are in internal equilibrium; see Section 3.8. A uniform body not in internal equilibrium with respect to a smaller set Z(t) of state variables can be made in internal equilibrium by extending the set of state variables. This justifies our claim in footnote 8. The vitrification process is reviewed in Section 4 where we introduce the concept of the residual thermodynamic entropy SR.
- We provide a first principles statistical formulation of nonequilibrium thermodynamic entropy for an isolated system in terms of microstate probabilities without the use of any dynamical laws36 in Section 5. The formulation is based on the Fundamental Axiom and is valid for any state of the body. We use a formal approach (frequentist interpretation of probability) by extending the equilibrium ensemble of Gibbs to a nonequilibrium ensemble, which is nothing but a large number of (mental) samples of the body under consideration; we refer the ensemble as a sample space. The formal approach enables us to evaluate the combinatorics for a given set of microstate probabilities. The resulting statistical entropy is independent of the number of samples and depends only on the probabilities as is seen from Equations (52) and (64). Thus, the use of a large number of samples is merely a mathematical formality and is not required in practice. We have shown that in equilibrium, the statistical entropy is the same as the equilibrium thermodynamic entropy: . However, we have also shown that the statistical entropy is equal to the thermodynamic entropy in IEQS described by the state variables . The IEQSs correspond to homogeneous states of the body. The conclusion also remains valid for an inhomogeneous system in ANEQS whose subsystems are individually homogeneous and in different IEQSs. We cannot make any comment about the relationship between and S(Z(t),t) for the simple reason that there is no way to measure or calculate a non-state function S(Z(t),t). Despite this, when the system is in a homogeneous ANEQS, the inequalities in Equations (56) and (34) still show the deep connection between the two entropies. Therefore, the statistical entropy in this case can be used to give the value of the corresponding thermodynamic entropy. This then finally allows us to make Conjecture 1 in Section 6. The discussion is extended to the case where state space is multi-component such as in glasses in Section 7.
- The second problem relates to the meaning of the number of microstates W that appears in the Boltzmann entropy. We have concluded in previous work [52,58] that temporal averages may not be relevant at low temperatures where component confinement occurs in of thermodynamically significant several disjoined components. The glass is formed when the system gets trapped in one of these components. Does W refer to the number of microstates within this component or to all microstates in all the components even though the glass is not allowed to probe these components over the observation period τobs < τeq? If dynamics is vital for the thermodynamics of the system, then it would appear that latter alternative should be unacceptable. Various reliable estimates for SR show that it is positive. Its positive value plays a very important role in clarifying the meaning of W. The answer is that W must contain all the microstates with non-zero probabilities in all disjoint components; see the Claim 6 in Section 7.
- In Section 8, we briefly review the semi-classical approximation for the entropy by introducing cells in phase space, whose volume is hs under adiabatic approximation. Using the cell construction, we show that Sf in Equation (22) cannot represent the entropy of the system. We argue that for nonequilibrium states, f may diverge as the cell volume dxc → 0. However, the product fdxc must be strictly ≤ 1. We consider a simple example of a 1-d damped oscillator for which we show that the above conclusion is indeed valid. Thus, contrary to what is commonly stated, e.g., [92,94], the divergence of f does not mean that the entropy also diverges.
- Thermodynamic entropy can become negative, but this is not possible for the statistical entropy. To clarify the issue, we consider a simple lattice model in Section 9, which in the continuum limit becomes the noninteracting Tonks gas. The latter model has been solved exactly and shows negative entropy at high coverage; see Equation (73). The entropy of the original lattice model is non-negative at all coverages and reduces to zero only when the coverage is full. It is found that one must subtract a term containing δ, the lattice spacing. By a proper subtraction, the lattice entropy has been shown to reproduce the continuum entropy in Equation (73) as δ → 0. It is a common practice to treat δ a constant, in which case the subtraction is harmless. However, there is no reason to assume δ to be state independent.
- One encounters inequalities such as in Equation (2), which creates problems in determining the entropy calorimetrically; the calorimetric determination of deQ and T0 is not sufficient to obtain dS. In our approach based on internal variables [38,39], see in particular the discussion starting below Equation (3) and leading to Equation (6), we have dQ = deQ + diQ = T (t)dS(t), dW = deW + diW = P(t)dV(t) +⋯+ A(t) · dξ(t) so that the inequality dS > deQ/T0 is replaced by an equality dS = dQ/T (t). Thus, both dQ and dW are expressed in terms of the fields of the body defined in Equation (35). This provides for tremendous simplification as internal equilibrium is very similar in many respect to equilibrium; see Section 3.6 and Ref. [38,39].
- For an isolated system in internal equilibrium (pα = p for ∀mα), just a single sample (or microstate) will suffice to determine the entropy as samples are unbiased. The entropy in this case is no different than the “entropy” − ln p of a single sample [52,55,56]:where W0 represents W0(Z0(t)) or W0(X0). The entropy of the “single” microstate in this case will always satisfy the second law for W0 continues to increase as the system relaxes.
- However, this simplicity is lost as soon as the system is not in internal equilibrium. Here, one must consider averaging over all microstates to evaluate the statistical entropy, which must satisfy the second law. In contrast, the negative of the index of probability −η(t) used in Equation (52), which has also been identified as the microstate entropy in modern literature [14–16], may increase or decrease with time. This is merely a consequence of probability conservation (see the second equation in Equation (49)), and is not related to second law, which refers to the behavior of the negative of the average index of probability.
- Some readers may think that our statistical formulation is no different than that used in the information theory. We disagree. For one, there is no concept of internal variables ξ0 in the latter theory. Because of this, our approach allows us to consider three levels of description so that we can consider three different entropies S0(Z0(t),t), S0(Z0(t)) and S0(X0) satisfying the inequalities in Equation (56). The information theory can only deal with two levels of entropies. There is also no possibility of a residual entropy in the latter.
- We should remark here that the standard approach to calculate nonequilibrium entropy is to use the classical nonequilibrium thermodynamics [3] or its variant, which treats the entropy at the local level as an equilibrium state function. Moreover, it can only treat an inhomogeneous system in a NEQS but not a homogeneous NEQS. Furthermore, the correlation length plays no role in determining the “elemental volume” over which local equilibrium holds [6] (p. 335). As no internal variables are required, it is different from our approach which exploits internal variables. Our approach is also different from those approaches in which the entropy is treated as a function of fluxes [9]. As fluxes depend strongly on how the system relates to the medium, they cannot serve as state variables for the system in our approach; see SP1 and the discussion following it.
Acknowledgments
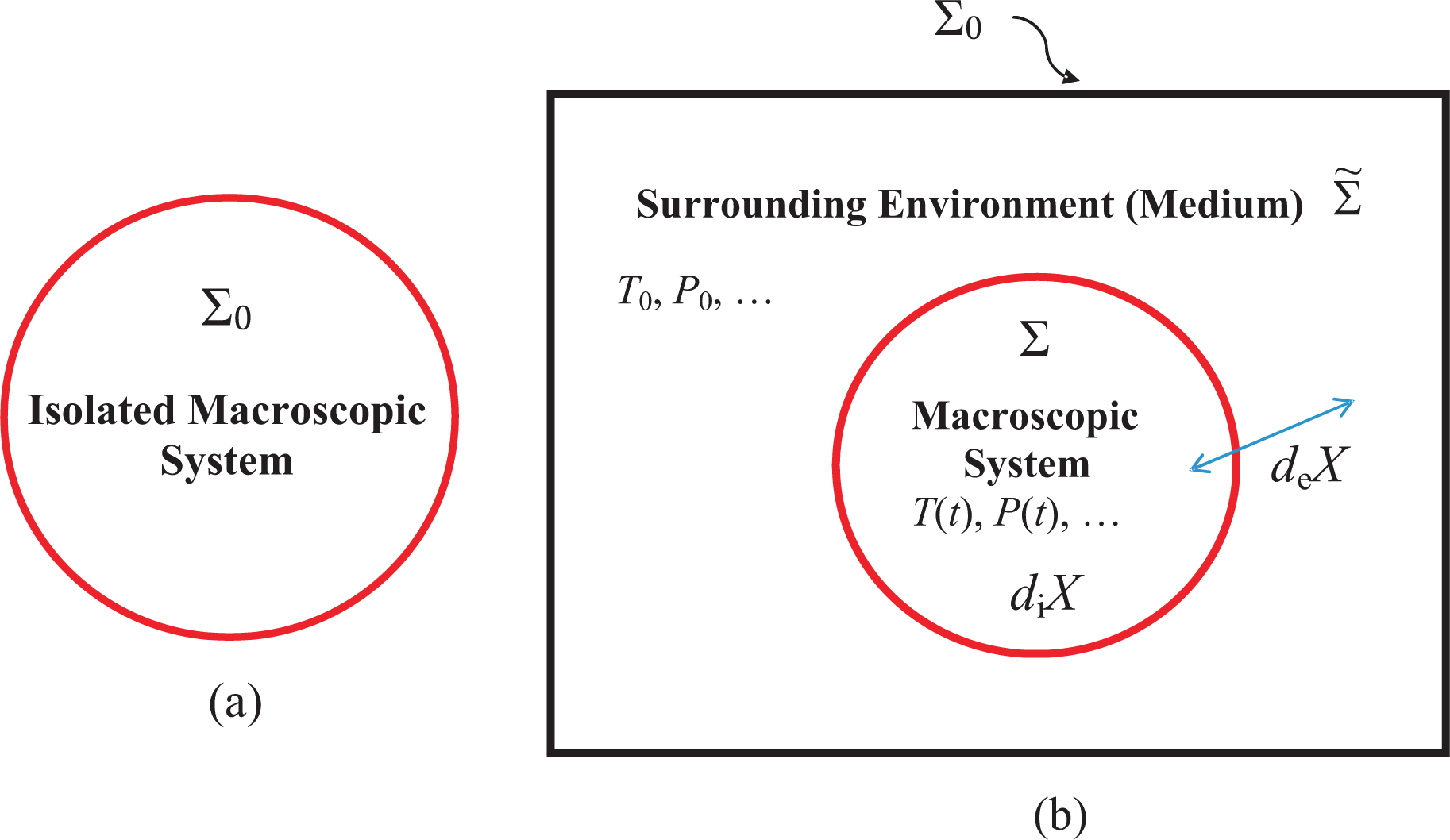
- 1The number of these variables is determined by the nature of description of the system appropriate in a given investigation of it. In equilibrium, the entropy is a unique function of these variables, called the observables. It should be noted that extensivity does not imply additivity (sum of parts equals the total) and vice versa. As additivity plays a central role in our approach, we will restrict ourselves to only short-ranged interactions or screened long-ranged interactions such as the electrostatic interactions screened over the Debye length [4] (Sections 78–80). The interaction in both cases is tempered and stable [40] (p. 320). In addition, we require our system to be mechanically and thermodynamically independent from the medium, see Figure 1b, in the sense discussed later in Section 3.8; however, see [5].
- 3The reversible entropy exchange deS always appears as an equality for any process (reversible or irreversible): deS = deQ/T0. In terms of the irreversible entropy diS ≥ 0 generated within the body, the entropy change is dS ≡ diS + deS, which then results in the Clausius inequality dS > deS for an irreversible process. Furthermore, we will reserve the use of dQ in this review to denote the sum dQ ≡ diQ + deQ, where diQ is irreversible heat generated within the system due to internal dissipation.
- 4Throughout this work, we only consider nonnegative temperatures of the medium.
- 5The energy can only change by exchanges with the medium. As no internal process can change the energy, we have diE = 0 so that dE = deE. To understand the nature of diX, we need to consider an isolated system. It is easy to see that diV = 0, but diW, diN, etc. need not be zero.
- 6Some of the physical significance of the entropy that will be discussed in the review are its identification as a macroscopic concept, its behavior in a spontaneous process, a possible state function, its relevance for thermodynamic forces and nonequilibrium thermodynamics, its equivalence with the statistical entropy that is always non-negative, its ability to explain the residual entropy, etc.
- 7The number of papers and books on nonequilibrium entropy is so vast that it is not possible to do justice to all of them in the review. Therefore, we limit ourselves to cite only a sample of papers and books that are relevant to this work and we ask forgiveness from authors who have been omitted.
- 8In Ref. [31], these authors suggest that the entropy, temperature, etc. for nonequilibrium states are approximate concepts. More recently [32], they have extended their approach to nonequilibrium systems but again refer to the limitation. We hope to show here that the situation is not so bleak and it is possible to make progress; see also [5].
- 9The subsystems cannot be truly independent if S has to change in time. Their quasi-independence will allow them to affect each other weakly to change S [4].
- 10Later we will see that this equivalence only holds true when all possible microstates, microstates that have nonzero probabilities, are counted regardless of whether they belong to disjoint components in (micro)state space (such as for glasses) or not.
- 11The observables are quantities that can be manipulated by an observer. It is a well-known fact from mechanics that the only independent mechanical quantities that are extensive are the energy and linear and angular momenta for a mechanical system [4] (see p. 11). To this, we add the volume and the number of particles of different species in thermodynamics. There may be other observables such as magnetization, polarization, etc. depending on the nature of the system. For an isolated system, all these observables are constant. In many cases, some of the extensive observables are replaced by their conjugate fields like temperature, pressure, etc. However, we find it useful to only use extensive observables.
- 12The set of all observables will be denoted by X0. For a body Σ, the set will be denoted by X. At least one (extensive) observable, to be denoted by X0f or Xf, must be held fixed to specify the macroscopic size of the body [59]. Usually, it is taken to be the number of particles N (of some pre-determined species if there are many species). Therefore, we must always set dX0f = 0 or dXf = 0 in those equations where it appears in this work.
- 14It is well known that the same formulation is applicable to any equilibrium ensemble.
- 15In other ensembles, the set will include all microstates corresponding to all possible values of fluctuating observables.
- 16This integral should not be confused with the integral along γ12 over which the second integral vanishes.
- 17These variables are also extensive just like the observables. However, they cannot be controlled by the observer; compare with the footnote 11.
- 18It must be remarked that a (thermodynamic) cyclic process is not possible for an isolated system. Such a system can only undergo a spontaneous process 0 → 0eq in Figure 2, which represents the spontaneous relaxation going on within Σ0 as the system tries to approach equilibrium. For Σ0 to return to 0 will violate the second law.
- 19If there is a repeating ANEQS for which its entropy has the repeating property S(Z(t + τC), t + τC) = S(Z(t), t) for a particular cycle time τC (of course, Z(t + τC) = Z(t)), one can construct a cyclic process with this state as the starting and the repeating state. It is clear that Equation (3) will also hold for such a cycle. We will not consider such a cyclic process here as it is a cycle only of a fixed cycle time.
- 21We assume that . The additivity of energy, a mechanical quantity, is therefore a consequence of a mechanical independence of subsystems.
- 22We assume that there is a single correlation length. If there are several correlation lengths, we take the longest of all for the discussion.
- 23We say that the two subsystems are thermodynamically independent as this independence is needed for S, which is a thermodynamic concept.
- 24We find it useful to think of as a function not only of the observables but also of internal variables in this section, even though the latter are not independent, so that we can later expand around Z0(t).
- 25Compare GX(t) with the concept of generalized available energy [5].
- 26The constraint is to disallow ordered configurations responsible for crystallization.
- 27It should be stressed that as it has not been possible to get to absolute zero, some extrapolation is always needed to determine Sexpt(0).
- 28It should be recalled that Boltzmann needed to assume molecular chaos to derive the second law; microscopic Newtonian dynamics of individual particles alone is not sufficient for the derivation; see for example [52] for justification, where it has been shown that microscopic Newtonian dynamics cannot bring about equilibration in an isolated body that is not initially in equilibrium. However, not everyone believes that the second law cannot be derived from Newtonian mechanics.
- 29The sets m and p should not be confused with the angular and linear momenta in the previous section.
- 30The entropy formulation in Equation (52) can include pα = 0 without any harm, even though it is preferable to not include it as the definition of the set m0(Z0) and the number W0(Z0) only includes microstates with non-zero probabilities.
- 31A thermodynamically significant number a is exponential (~ eαN) in the number of particles N with α > 0. This ensures that as N → ∞. One can also consider the limiting form of a as α → 0 from above.
- 32The adiabatic approximation (very slow variation of parameters in time) in mechanics [90] should not be confused with thermodynamic adiabatic approximation for which deQ = 0.
- 33The partition function Z should not be confused with the state variable Z.
- 34The enrgy E contains only the kinetic energy of translation, which determines the temperature. However, the kinetic energy is of no concern in the follwing for studying the configurational entropy sc.
- 35Observe that we do not consider S/N or S/Nf as these quantities are ambiguous because the denomenators may depend on the state but also because they vanish in the limit.
References and Notes
- Clausius, R. The Mechanical Theory of Heat; Macmillan & Co.: London, UK, 1879. [Google Scholar]
- De Donder, Th.; van Rysselberghe, P. Thermodynamic Theory of Affinity; Stanford University: Stanford, CA, USA, 1936. [Google Scholar]
- De Groot, S.R.; Mazur, P. Non-Equilibrium Thermodynamics, 1st ed; Dover: New York, NY, USA, 1984. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Statistical Physics, 3rd ed; Pergamon Press: Oxford, UK, 1986; Volume 1. [Google Scholar]
- Gyftopoulos, E.P.; Beretta, G.P. Thermodynamics Foundations and Application; Macmillan Publishing Company: New York, NY, USA, 1991. [Google Scholar]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics; John Wiley and Sons: West Sussex, UK, 1998. [Google Scholar]
- Öttinger, H.C. Beyond Equilibrium Thermodynamics; Wiley-Interscience: Hoboken, NJ, USA, 2005. [Google Scholar]
- Mueller, I.; Ruggeri, T. Rational Extended Thermodynamics, 2nd ed; Springer-Verlag: New York, NY, USA, 1998. [Google Scholar]
- Lebon, G.; Joue, D.; Casas-Vásgues, J. Understanding Non-equilibrium Thermodynamics; Springer-Verlag: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Gibbs, J.W. Elementary Principles in Statistical Mechanics; Yale University Press: New Haven, CT, USA, 1960. [Google Scholar]
- Tolman, R.C. The Principles of Statistical Mechanics; Oxford University: London, UK, 1959. [Google Scholar]
- Rice, S.A.; Gray, P. The Statistical Mechanics of Simple Liquids; Interscience Publishers: New York, NY, USA, 1965. [Google Scholar]
- Keizer, J. Statistical Thermodynamics of Nonequilibrium Processes; Springer-Verlag: New York, NY, USA, 1987. [Google Scholar]
- Campisi, M.; Hänggi, P.; Talkner, P. Quantum fluctuation relations: Foundations and applications. Rev. Mod. Phys. 2011, 83, 771–791. [Google Scholar]
- Siefert, U. Stochastic thermodynamics: principles and perspectives. Eur. Phys. J. B. 2008, 64, 423–431. [Google Scholar]
- Gawedzki, K. Fluctuation Relations in Stochastic Thermodynamics 2013, arXiv, 1308.1518v1.
- Landau, L.D.; Lifshitz, E.M. Quantum Mechanics, 3rd ed; Pergamon Press: Oxford, UK, 1977. [Google Scholar]
- Von Neumann, J. Mathematical Foundations of Quantum Mechanics; Princeton University Press: Princeton, NJ, USA, 1996. [Google Scholar]
- Partovi, M.H. Entropic Formulation of Uncertainty for Quantum Measurements. Phys. Rev. Lett. 1983, 50, 1883–1885. [Google Scholar]
- Bender, C.M.; Brody, D.C.; Meister, B.K. Quantum mechanical Carnot engine. J. Phys. A. 2000, 33, 4427–4436. [Google Scholar]
- Unusual quantum states: Non–locality, entropy, Maxwell’s demon and fractals. Proc. R. Soc. A. 2005, 461, 733–753.
- Scully, M.O.; Zubairy, M.S.; Agarwal, G.S.; Walther, H. Extracting Work from a Single Heat Bath via Vanishing Quantum Coherence. Science 2003, 299, 862–864. [Google Scholar]
- Beckenstein, J.D. Black Holes and Entropy. Phys. Rev. D. 1973, 7, 2333–2346. [Google Scholar]
- Beckenstein, J.D. Statistical black-hole thermodynamics, ibid. Phys. Rev. D 1975, 12, 3077–3085. [Google Scholar]
- Schumacker, B. Quantum coding. Phys. Rev. A. 1995, 51, 2738–2747. [Google Scholar]
- Bennet, C.H. The Thermodynamics of Computation—A Review. Int. J. Theor. Phys. 1982, 21, 905–940. [Google Scholar]
- Bennet, C.H. Quantum information. Phys. Scr. 1998, T76, 210–217. [Google Scholar]
- Wiener, N. Cybernetics; MIT Press: Cambridge, UK, 1948. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar]
- Balian, R. Entropy, a Protean Concept. Poincaré Semianr 2003, 2, 119–145. [Google Scholar]
- Lieb, E.H.; Yngvason, J. The physics and mathematics of the second law of thermodynamics. Phys. Rep. 1999, 310, 1–96. [Google Scholar]
- Lieb, E.H.; Yngvason, J. The entropy concept for non-equilibrium states 2013, arXiv, 1305.3912.
- Gyftopoulos, E.P.; Çubukçu, E. Entropy: Thermodynamic definition and quantum expression. Phys. Rev. E. 1997, 55, 3851–3858. [Google Scholar]
- Beretta, G.P.; Zanchini, E. A definition of thermodynamic entropy valid for non-equilibrium states and few-particle systems 2014, arXiv, 1411.5395.
- Gujrati, P.D. Non-equilibrium Thermodynamics: Structural Relaxation, Fictive temperature and Tool-Narayanaswamy phenomenology in Glasses. Phys. Rev. E. 2010, 81, 051130. [Google Scholar]
- Gujrati, P.D. Nonequilibrium thermodynamics. II. Application to inhomogeneous systems. Phys. Rev. E. 2012, 85, 041128. [Google Scholar]
- Gujrati, P.D.; Aung, P.P. Nonequilibrium thermodynamics. III. Generalization of Maxwell, Clausius-Clapeyron, and response-function relations, and the Prigogine-Defay ratio for systems in internal equilibrium. Phys. Rev. E. 2012, 85, 041129. [Google Scholar]
- Gujrati, P.D. Generalized Non-equilibrium Heat and Work and the Fate of the Clausius Inequality 2011, arXiv, 1105.5549.
- Gujrati, P.D. Nonequilibrium Thermodynamics. Symmetric and Unique Formulation of the First Law, Statistical Definition of Heat and Work, Adiabatic Theorem and the Fate of the Clausius Inequality: A Microscopic View 2012, arXiv, 1206.0702. [Google Scholar]
- Lieb, E.H.; Lebowitz, J.L. The constitution of matter: Existence of thermodynamics for systems composed of electrons and nuclei. Adv. Math. 1972, 9, 316–398. [Google Scholar]
- Gallavotti, G. Entropy production in nonequilibrium thermodynamics: A review 2004, arXiv, cond-mat/0312657v2.
- Ruelle, D. Positivity of entropy production in nonequilibrium statistical mechanics. J. Stat. Phys. 1996, 85, 1–25. [Google Scholar]
- Ruelle, D. Extending the definition of entropy to nonequilibrium steady states. Proc. Natl. Acad. Sci. 2003, 85, 3054–3058. [Google Scholar]
- Oono, Y.; Paniconi, M. Steady state thermodynamics. Prog. Theor. Phys. 1998, 30, 29–44. [Google Scholar]
- Maugin, G.A. The Thermomechanics of Nonlinear Irreversible Behaviors: An Introduction; World Scientific: Singapore, Singapore, 1999. [Google Scholar]
- Beretta, G.P.; Zanchini, E. Rigorous and General Definition of Thermodynamic Entropy. In Thermodynamics; Tadashi, M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 23–50. [Google Scholar]
- Canessa, E. Oscillating Entropy 2013, arXiv, 1307.6681.
- Sasa, S. Possible extended forms of thermodynamic entropy 2013, arXiv, 1309.7131.
- Beretta, G.P.; Zanchini, E. Removing Heat and Conceptual Loops from the Definition of Entropy. Int. J. Thermodyn. 2010, 12, 67–76. [Google Scholar]
- Feynman, R.P. The Feynman Lectures on Physics; Addison-Wesley: Boston, MA, USA, 1963; Volume 1. [Google Scholar]
- Zanchini, E.; Beretta, G.P. Recent Progress in the Definition of Thermodynamic Entropy. Entropy 2014, 16, 1547–1570. [Google Scholar]
- Gujrati, P.D. Loss of Temporal Homogeneity and Symmetry in Statistical Systems: Deterministic Versus Stochastic Dynamics. Symmetry 2010, 2, 1201–1249. [Google Scholar]
- Bishop, R.C. Nonequilibrium Statistical Mechanics Brussels-Austin. Style. Stud. Hist. Philos. Mod. Phys. 2004, 35, 1–30. [Google Scholar]
- Lavis, D.A. Boltzmann, Gibbs, and the Concept of Equilibrium. Philos. Sci. 2008, 75, 682–696. [Google Scholar]
- Lebowitz, J. Statistical mechanics: A selective review of two central issues. Rev. Mod. Phys. 1999, 71, S346–S357. [Google Scholar]
- Goldstein, S.; Lebowitz, J.L. On the (Boltzmann) Entropy of Nonequilibrium Systems 2003, arXiv, cond-mat/0304251.
- Palmer, R.G. Broken ergodicity. Adv. Phys. 1982, 31, 669–735. [Google Scholar]
- Gujrati, P.D. Energy gap Model of Glass Formers: Lessons Learned from Polymers. In Modeling and Sinulation in Polymers; Gujrati, P.D., Leonov, A.I., Eds.; Wiley-VCH: Weinheim, Germany, 2010; pp. 433–495. [Google Scholar]
- Gujrati, P.D. General theory of statistical fluctuations with applications to metastable states, Nernst points, and compressible multi-component mixtures. Recent Res. Devel. Chem. Phys. 2003, 4, 243–275. [Google Scholar]
- Planck, M. Über das Gesetz der Energieverteilung im Normalspektrum. Ann. Phys. 1901, 4, 553–563. [Google Scholar]
- Boltzmann, L. Über die Beziehung zwischen dem zweiten Hauptsatze der mechanischen Wärmtheorie und der Wahrscheinlichkeitscrechnung respektive den Sätzen über das Wärmegleichgewicht. Wien Ber 1877, 76, 373–435. [Google Scholar]
- Boltzman, L. Lectures on Gas Theory; University of California Press: Berkeley, CA, USA, 1964; pp. 55–62. [Google Scholar]
- The number of combinations in Equation (35) on p. 56 in Boltzmann [62] is denoted by Z, but it is not the number of microstates. The two become the same only when Z is maximized as discussed on p. 58
- Jaynes, E.T. Gibbs vs. Boltzmann Entropies. Am. J. Phys. 1965, 33, 391–398. [Google Scholar]
- Cohen, E.G.D. Einstein and Boltzmann: Determinism and Probability or The Virial Expansion Revisited 2013, arXiv, 1302.2084.
- Pokrovskii, V.N. A Derivation of the Main Relations of Nonequilibrioum Thermodynamics. ISRN Thermodyn 2013. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifshitz, E.M. Fluid Mechanics; Pergamon Press: Oxford, UK, 1982. [Google Scholar]
- Edwards, S.F.; Oakeshott, R.S.B. Theory of Powders. Physica 1989, 157A, 1080–1090. [Google Scholar]
- Bouchbinder, E.; Langer, J.S. Nonequilibrium thermodynamics of driven amorphous materials. I. Internal degrees of freedom and volume deformation. Phys. Rev. E. 2009, 80, 031131. [Google Scholar]
- Gutzow, I.; Schmelzer, J. The Vitreous State Thermodynamics, Structure, Rheology and Crystallization; Springer-Verlag: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Nemilov, S.V. Thermodynamic and Kinetic Aspects of the Vitreous State; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Gujrati, P.D. Where is the residual entropy of a glass hiding? 2009, arXiv, 0908.1075.
- Jäckle, J. On the glass transition and the residual entropy of glasses. Philos. Mag. B. 1981, 44, 533–545. [Google Scholar]
- Jäckle, J. Residual entropy in glasses and spin glasses. Physica B 1984, 127, 79–86. [Google Scholar]
- Gibson, G.E.; Giauque, W.F. The third law of thermodynamics. evidence from the specific heats of glycerol that the entropy of a glass exceeds that of a crystal at the absolute zero. J. Am. Chem. Soc. 1923, 45, 93–104. [Google Scholar]
- Giauque, W.E.F.; Ashley, M. Molecular Rotation in Ice at 10◦K. Free Energy of Formation and Entropy of Water. Phys. Rev. 1933, 43, 81–82. [Google Scholar]
- Pauling, L. The Structure and Entropy of Ice and of Other Crystals with Some Randomness of Atomic Arrangement. J. Am. Chem. Soc. 1935, 57, 2680–2684. [Google Scholar]
- Bestul, A.B.; Chang, S.S. Limits on Calorimetric Residual Entropies of Glasses. J. Chem. Phys. 1965, 43, 4532–4533. [Google Scholar]
- Nagle, J.F. Lattice Statistics of Hydrogen Bonded Crystals. I. The Residual Entropy of Ice. J. Math. Phys. 1966, 7, 1484–1491. [Google Scholar]
- Bowles, R.K.; Speedy, R.J. The vapour pressure of glassy crystals of dimers. Mole. Phys. 1996, 87, 1349–1361. [Google Scholar]
- Isakov, S.V.; Raman, K.S.; Moessner, R.; Sondhi, S.L. Magnetization curve of spin ice in a [111] magnetic field. Phys. Rev. B. 2004, 70, 104418. [Google Scholar]
- Berg, B.A.; Muguruma, C.; Okamoto, Y. Residual entropy of ordinary ice from multicanonical simulations. Phys. Rev. B. 2007, 75, 092202. [Google Scholar]
- Gujrati, P.D. Poincare Recurrence, Zermelo’s Second Law Paradox, and Probabilistic Origin in Statistical Mechanics 2008, arXiv, 0803.0983.
- Searles, D.; Evans, D. Fluctuations relations for nonequilibrium systems. Aust. J. Chem. 2004, 57, 1119–1123. [Google Scholar]
- Jaynes, E.T. Information Theory and Statistical Mechanics. Phys. Rev. 1957, 106, 620–630. [Google Scholar]
- Jaynes, E.T. Information Theory and Statistical Mechanics. II, ibid. Phys. Rev. 1957, 108, 171–190. [Google Scholar]
- Jaynes, E.T. Prior Probabilities. IEEE Trans. Syst. Sci. Cybern. 1968, 4, 227–241. [Google Scholar]
- Jaynes, E.T. Probability Theory: The logic of Science; Cambridge University Press: New York, NY, USA, 2003. [Google Scholar]
- Bagci, G.B.; Oikonomou, T.; Tirnakli, U. Comment on “Essential discreteness in generalized thermostatistics with non-logarithmic entropy” by S. Abe 2010, arXiv, 1006.1284v2.
- Landau, L.D.; Lifshitz, E.M. Mechanics, 3rd ed; Pergamon Press: Oxford, UK, 1976. [Google Scholar]
- Holian, B.L. Entropy evolution as a guide for replacing the Liouville equation. Phys. Rev. A. 1986, 34, 4238–4245. [Google Scholar]
- Holian, B.L.; Hoover, W.G.; Posch, H.A. Resolution of Loschmidt’s paradox: The origin of irreversible behavior in reversible atomistic dynamics. Phys. Rev. Lett. 1987, 59, 10–13. [Google Scholar]
- Ramshaw, J.D. Remarks on entropy and irreversibility in non-hamiltonian systems. Phys. Lett. A. 1986, 116, 110–114. [Google Scholar]
- Hoover, W.G. Liouville’s theorems, Gibbs’ entropy, and multifractal distributions for nonequilibrium steady states. J. Chem. Phys. 1998, 109, 4164–4170. [Google Scholar]
- Semerianov, F.; Gujrati, P.D. Configurational entropy and its crisis in metastable states: Ideal glass transition in a dimer model as a paragidm of a molecular glass. Phys. Rev. E. 2005. [Google Scholar] [CrossRef]
- Tonks, L. The Complete Equation of State of One, Two and Three-Dimensional Gases of Hard Elastic Spheres. Phys. Rev. 1936, 50, 955–963. [Google Scholar]
- Thompson, C. Mathematical Statistical Mechanics; Princeton University Press: Princeton, NJ, USA, 1972. [Google Scholar]
- Gujrati, P.D. A binary mixture of monodisperse polymers of fixed architectures, and the critical and the theta states. J. Chem. Phys. 1998, 108, 5104–5121. [Google Scholar]
- Hatsopoulos, G.N.; Gyftopoulos, E.P. A unified quantum theory of mechanics and thermodynamics, Part I. Postulates Found. Phys. 1976, 6, 15–31. [Google Scholar]
- Beretta, G.P. Quantum Thermodynamics of Non-Equilibrium. Onsager Reciprocity and Dispersion-Dissipation Relations. Found. Phys. 1987, 17, 365–381. [Google Scholar]




© 2015 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gujrati, P.D. On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies. Entropy 2015, 17, 710-754. https://doi.org/10.3390/e17020710
Gujrati PD. On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies. Entropy. 2015; 17(2):710-754. https://doi.org/10.3390/e17020710
Chicago/Turabian StyleGujrati, Purushottam D. 2015. "On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies" Entropy 17, no. 2: 710-754. https://doi.org/10.3390/e17020710
APA StyleGujrati, P. D. (2015). On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies. Entropy, 17(2), 710-754. https://doi.org/10.3390/e17020710