1. Introduction
Since the pioneering work of Aviram and Ratner [
1] discussing the use of a single organic molecule to construct a rectifier, molecular electronics has been a continuously expanding field [
2]. A number of applications have been developed, such as the organic synthesis of conductive polymers [
3]. In the present paper, we discuss a possible application of a curious property of aromaticity, the existence of a small negative contribution of aromaticity to entropy.
It is well known that aromatic compounds present much lower enthalpy of formation than the one expected for the corresponding Kekule structures. Similarly, more than sixty years ago, Franklin [
4] predicted a weak and negative contribution of aromaticity to entropy. An example of this universal property is the adsorption of pyridine on Si(100): experimental results [
5,
6] showed that dative attachment which is the only way to preserve aromaticity is the favored form of adsorbed pyridine in dilute solutions at low temperatures. Recent theoretical analysis [
7,
8,
9,
10] confirmed that dative configuration is the more stable configuration. Two factors limit the domain of stability of dative attachment: repulsive interactions between dative bonds prevent them from being present in concentrated solutions while aromaticity contributes to a decrease in the entropy. When heated, aromatic grafted species (dative bonded) are converted to non-aromatic species which explains the vanishing of dative bonds at high temperatures even in dilute solutions as experimentally observed [
5,
6]. Since aromatic configuration is favored by lower enthalpy, this conversion appears driven by the entropic gain resulting from the vanishing of aromaticity [
7,
8] justifying here the deliberate use of possible negative contribution of aromaticity to entropy.
Recently, Casati et al. [
11] show that when pressure is applied in aromatic compounds molecular stability which originates from the resonance between electronic configurations can be decreased in molecular crystals that gradually localizes one of the resonant configurations on compression.
This paper which is devoted to thermal switches used for circuit protection aims to go one step further by considering the combined effect of the temperature in addition to compression. Generally, thermal switches rely on a sharp drop in conductivity at the switch temperature, typically in the 300–500 K range. In the following, we shall discuss how the transition from an aromatic structure towards a non-aromatic structure can be leveraged to build molecular junctions for use as thermal switches. The negative contribution of aromaticity to entropy allows the existence of a transition temperature at which the entropy contribution compensates the energy contribution. Moreover, aromaticity is related to the cyclic delocalized distribution of π electrons [
12]. Then, it may be expected that conductivity drops when aromaticity is lost at the transition temperature [
13].
2. Thermal Switches
In order to discuss the relevance of this approach, let us use the results of earlier studies [
7,
8] for pyridine adsorbed on Si(100) and Ge(100), even if these materials are certainly not the easiest ones to handle. Of particular interest for our purpose is the spectroscopic study associated to DFT calculations of gold-molecule-gold junctions made by Yoshida et al. [
14]. These authors have studied symmetric tolane-type molecules with pyridil as the anchoring group. Instead of gold, let us consider a tolane derived junction NC
5H
4–C≡C–C
5H
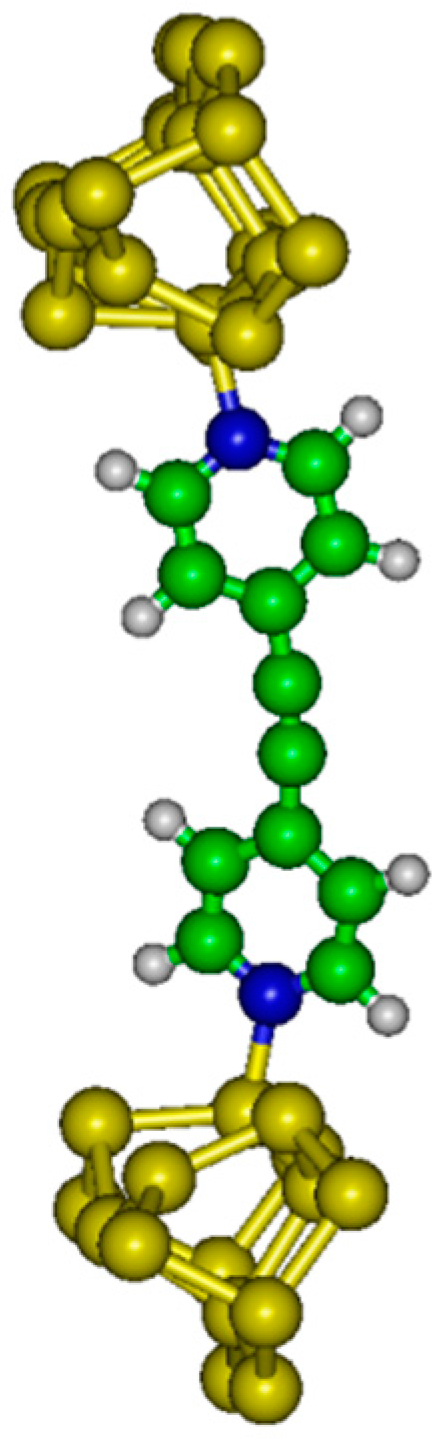
4N between two doped silicon samples. Two possible molecular devices are considered in the following: molecular junction with dative–dative (aromatic) attachment to Si(100) electrodes (see
Figure 1) or dative–tetrapod (non-aromatic) attachment (see
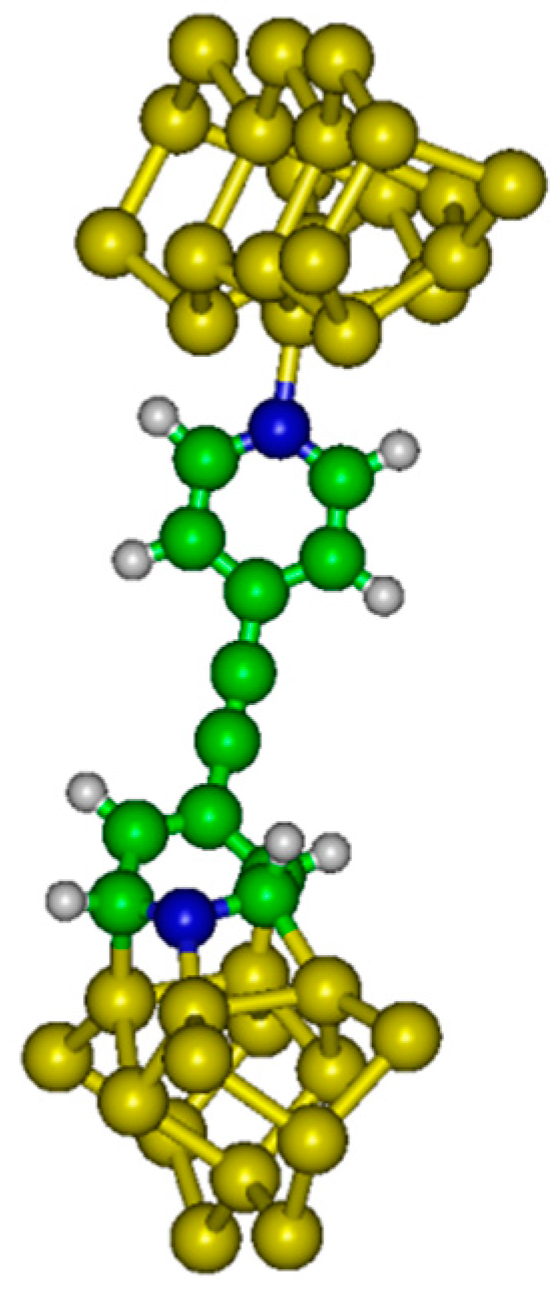
Figure 2).
3. Temperature Dependence of the Structure
It is of interest to plot the free energy (F = U − T·S) as a function of the distance between electrodes in both dative–dative and dative–tetrapod configurations.
The internal energy (U) has been evaluated as a function of the distance between electrodes (h). Electronic structure calculations have been completed with the GAMESS (US) software package. In the system considered here, the molecule is grafted between two semi-conducting silicon electrodes.
A Si
21H
20 cluster has been used to model the up electrode (with dative attachment). The top six silicon atoms compose the surface dimers while the remaining 15 Si atoms compose two subsurface layers which are hydrogen terminated to preserve the sp
3 hybridization of the bulk diamond lattice. A Si
15H
18 cluster has been used to model the bottom electrode (with dative or tetrapod attachment). The top four Si atoms compose the surface dimers while the remaining eleven Si atoms compose two subsurface layers. We have considered different altitudes
h (see
Figure 3) between the two silicon electrodes in the range 19–22 Å.
Geometries of local minima on the potential energy surface were calculated at DFT (B3LYP) level of theory, with a 6–31 G* basis set for nitrogen, carbon and terminating hydrogens and a SBKJC pseudo-potential set for Si atoms. The geometry optimization consists to freeze the last silicon row of each electrode with full optimization of all the other atomic positions.
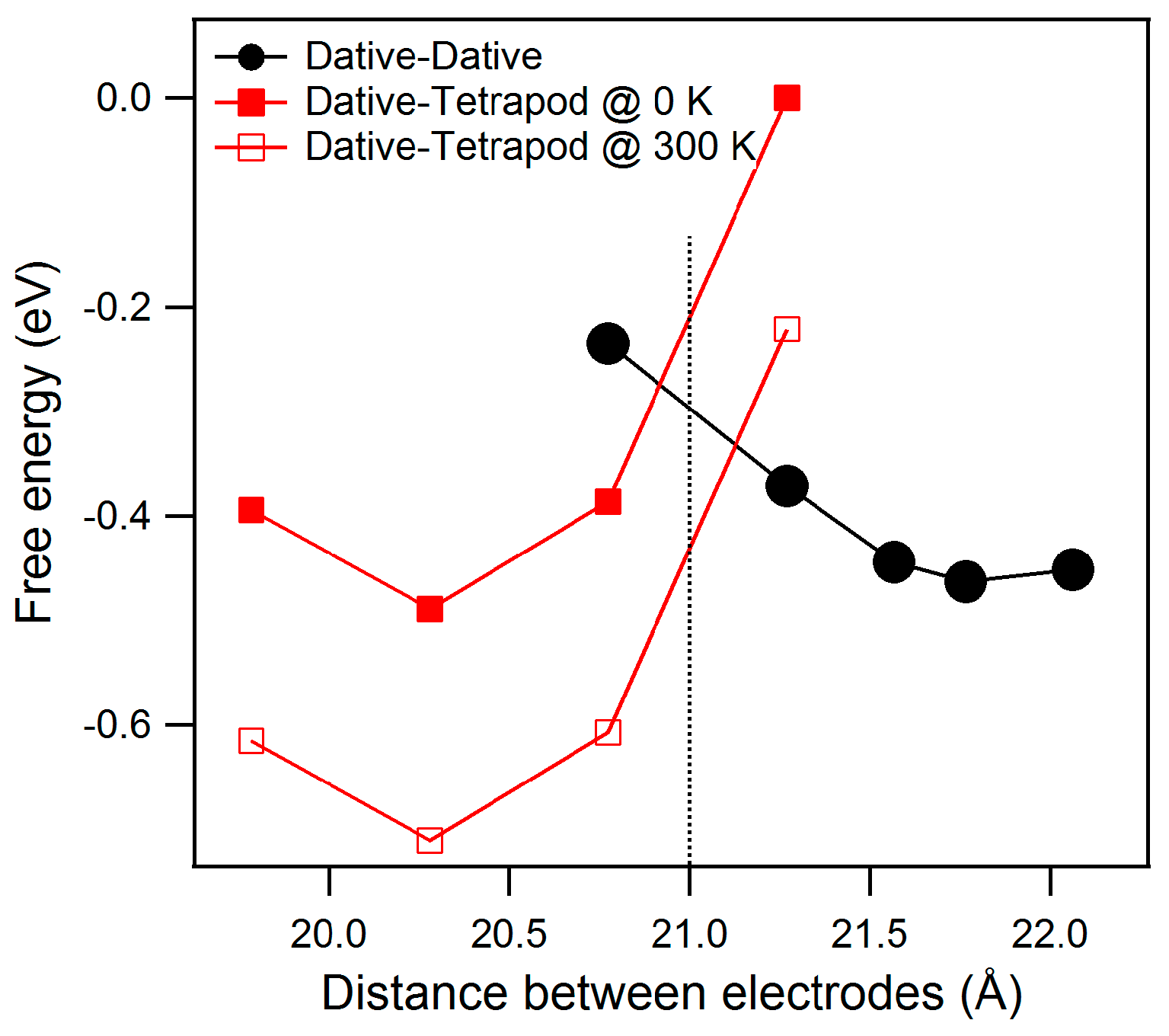
Our DFT simulations (see
Figure 3) provide an estimate of the energy (the free energy at low temperature) for both configurations as function of
h.
For large values of h, the dative–dative configuration is the most stable form. A minimum (EDD) of energy is found at h = 21.7 Å. At weaker altitudes the chain transforms to the dative–tetrapod configuration, with a minimum (EDT) of energy calculated at 20.3 Å.
It is interesting to note that both configuration present the same energy for h close to 21 Å.
The main point is: because of the dative–dative chain is longer than the dative–tetrapod one, an intermediate altitude at which both energy contributions are equal can be inferred. Our simplistic approach (zero point energy or extreme low-temperature conductivity effects are ignored here) gives an estimate of this altitude: knowledge of the exact value is not needed to further discuss their relative stability.
We assume the difference in entropy between aromatic (dative) and non-aromatic (tetrapod) configurations to be the one evaluated for pyridine in our earlier study [
7,
8] i.e., 17 cal·mol
−1·K
−1.
It is noteworthy that this small entropy difference is able to reverse the previous prediction derived from the energy difference alone. At moderate temperature (e.g., 300 K) the entropic term (−T·ΔS) is significant with respect to ΔU and, at fixed
h = 21 Å (then fixed volume), the dative–tetrapod configuration is favored (see
Figure 3).
This change of conformation of grafted molecule may have some consequences onto the local electronic conductivity across the molecular chain.
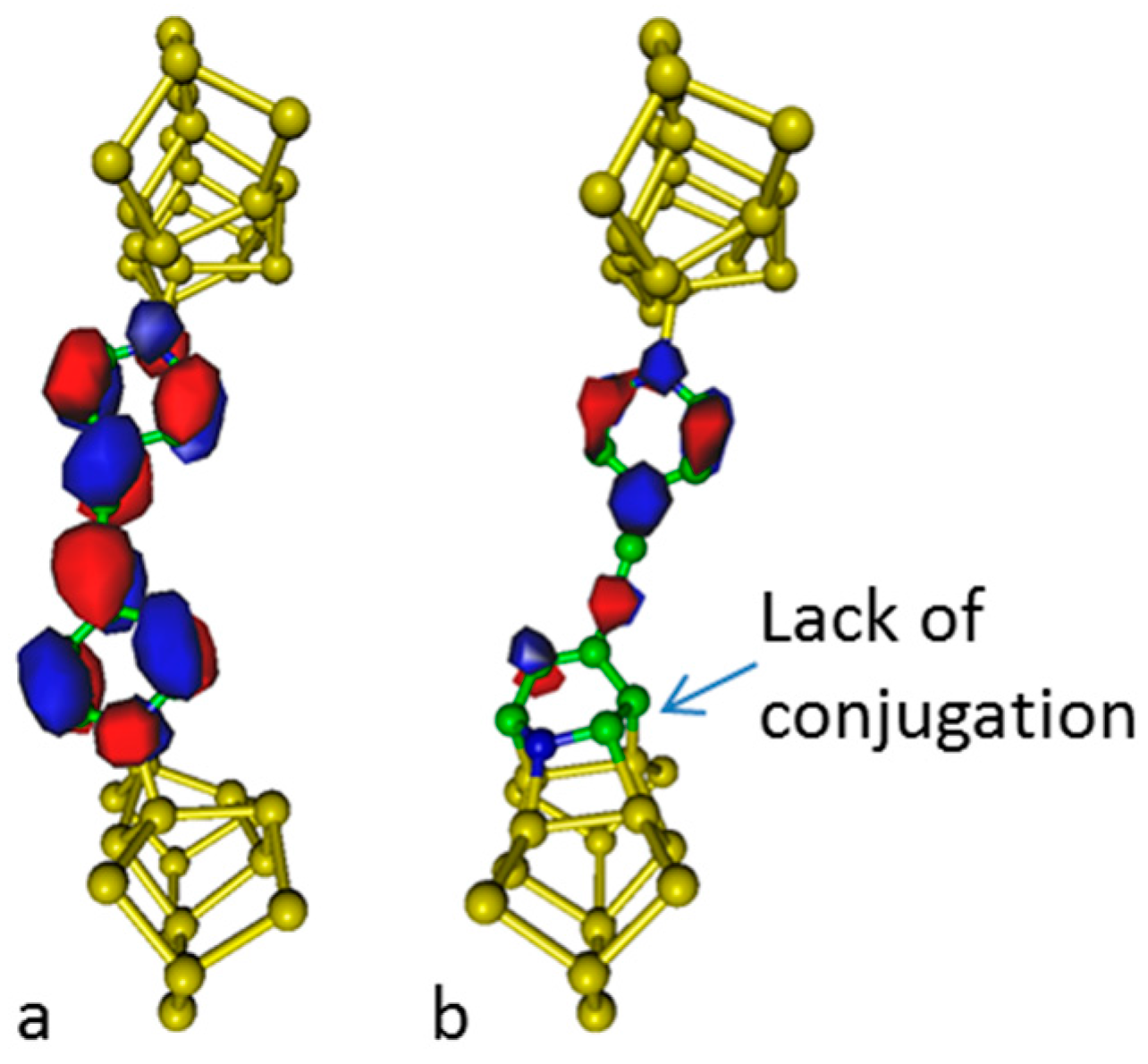
Analysis of the electronic density of the lowest unoccupied molecular orbital delocalized along the molecular chain is given in
Figure 4. It appears that the conjugation within the molecular junction vanishes as the dative pattern is replaced by a tetrapod pattern and then, a change in conductivity is expected. At high temperature, the negative contribution of aromaticity to entropy favors non-aromatic structures; this property may be used to build thermal switches.
4. Summary and Conclusions
Let us briefly summarize our approach restricted to dative–dative and dative–tetrapod configurations of NC5H4–C≡C–C5H4N junction between two Si(100) electrodes. The key quantity is the difference in free energy, ΔF = ΔU − T·ΔS, between aromatic and non-aromatic forms. ΔU and ΔS were determined separately.
ΔU was evaluated from ab-initio (DFT) calculations. At large distances (h) between Si electrodes the dative–dative configuration is the most stable form. At weaker distances the dative–tetrapod becomes the most stable form. For a distance h between Si electrodes close to 21 Å the energy of the dative–dative configuration is found very close to the one of the dative–tetrapod configuration.
ΔS was assumed to be close to the contribution of aromaticity to entropy deduced for pyridine from thermodynamical measurements [
7,
8].
Then, the sign of ΔF was used to qualitatively predict the stabilization of dative–tetrapod configuration when temperature increases making possible to build thermal switches using the small negative contribution of aromaticity to entropy.
Acknowledgments
We gratefully acknowledge Y-Lan Boureau and Jean-Michel Mariot for a critical reading of the manuscript.
Author Contributions
All authors have read and approved the final manuscript; the authors contributed equally to this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Chem. Phys. Lett. 1974, 29, 277–283. [Google Scholar] [CrossRef]
- Ratner, M. A brief history of molecular electronics. Nat. Nanotechnol. 2013, 8, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Akamatu, H.; Inokuchi, H.; Matsunaga, Y. Electrical Conductivity of the Perylene–Bromine Complex. Nature 1954, 173, 168–169. [Google Scholar] [CrossRef]
- Franklin, J.L. Calculation of Resonance Energies. J. Am. Chem. Soc. 1950, 72, 4278–4280. [Google Scholar] [CrossRef]
- Tao, F.; Qiao, M.H.; Wang, Z.H.; Xu, G.Q. Dative and Di−σ Binding States of Pyridine on Si(100) and Their Thermal Stability. J. Phys. Chem. B 2003, 107, 6384–6390. [Google Scholar] [CrossRef]
- Coustel, R.; Carniato, S.; Rochet, F.; Witkowski, N. Pyridine on Si(001)-(2×1): Density functional theory simulations compared with spectroscopic measurements. Phys. Rev. B 2012, 85, 035323. [Google Scholar] [CrossRef]
- Coustel, R.; Carniato, S.; Boureau, G. Thermodynamic factors limiting the preservation of aromaticity of adsorbed organic compounds on Si(100): Example of the pyridine. J. Chem. Phys. 2011, 134, 234708. [Google Scholar] [CrossRef] [PubMed]
- Coustel, R.; Carniato, S.; Boureau, G. Entropy, the Silent Killer of Aromaticity of Adsorbed Pyridine on Si(100) and Ge(100). J. Phys. Chem. C 2014, 118, 17505–17510. [Google Scholar] [CrossRef]
- Romeo, M.; Balducci, G.; Stener, M.; Fronzoni, G. N1s and C1s Near-Edge X-ray Absorption Fine Structure Spectra of Model Systems for Pyridine on Si(100): A DFT Simulation. J. Phys. Chem. C 2014, 118, 1049–1061. [Google Scholar] [CrossRef]
- Ng, W.K.H.; Liu, J.W.; Liu, Z.-F. Reaction Barriers and Cooperative Effects for the Adsorption of Pyridine on Si(100). J. Phys. Chem. C 2013, 117, 26644–26651. [Google Scholar] [CrossRef]
- Casati, N.; Kleppe, A.; Jephcoat, A.P.; Macchi, P. Putting pressure on aromaticity along with in situ experimental electron density of a molecular crystal. Nat. Commun. 2016, 7, 10901. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef] [PubMed]
- Wudl, F. Is there a relationship between aromaticity and conductivity? Pure Appl. Chem. 1982, 54, 1051–1058. [Google Scholar] [CrossRef]
- Yoshida, K.; Pobelov, I.V.; Manrique, D.Z.; Pope, T.; Mészáros, G.; Gulcur, M.; Bryce, M.R.; Lambert, C.J.; Wandlowski, T. Correlation of breaking forces, conductances and geometries of molecular junctions. Sci. Rep. 2015, 5, 9002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).


{kind=link}
{kind=link}
{kind=link}
{kind=link}



