2. Two Necessary Primitives: Energy and Exergy
Consider an arbitrary system
P of mass
M known to be in a state
P(
t = 0) of non-equilibrium. If the analysis addresses only scales sufficiently removed from the smallest ones (those identified by statistical mechanics behavior), and if the gradients of the relevant measurables remain bounded throughout the system, we can divide
P(
t = 0) in a finite number
k of spatial domains
δPj such that
, each one small enough to be considered in local equilibrium within the timescales imposed by the evolution of
P: notice that this assumption, often criticized by theoreticians as lacking of rigor for its being clearly an approximation of reality, is currently a standard and very successful procedure adopted in numerical calculations of structural, thermal and fluid-dynamic processes (finite-elements, finite-volumes techniques). Each one of these domains has a measurable energy (
cpjmjTj if thermal, 0.5
mjVj2 if kinetic, etc.) and thus both the total and the specific energy of
P at the time we open our window of observation are computable as well:
At any other instant
τ, if it is possible to identify another finite number
m, not necessarily equal to
k, of spatial domains, each one still small enough to be considered in local equilibrium within the prevailing timescales between
t = 0 and
t = τ, the energy of the (closed) system is:
Since the time interval is arbitrary, as long as the above formulated local equilibrium assumption is acceptable, the evolution of the energy of the system is:
where
dU/dt denotes the “infinitesimal” (continuous or finite) variation in time of the system’s energy.
If P is also isolated, then U(t) = U(0) at any time, and the evolution shall consist of a redistribution of the energy among the small spatial subdomains in which P is divided at each time. If P can exchange energy with the external world, then dU/dt is dictated by the prevailing boundary conditions.
3. Availability and Exergy
According to the original definition by Gibbs [
20], the
available energy or
availability A is a thermodynamic function representing the maximum work that can be extracted from a system that proceeds from an initial arbitrary state to its final “internal” equilibrium state (i.e., the one attained while isolated from its surroundings). No restraint is imposed on the initial state of the system, and no interaction with the environment is postulated except for an ideal “available energy” release [
21,
22]. Under the assumptions posited in the previous section, each subdomain
j of
P has a perfectly computable (or measurable) available energy. Since the final state is an equilibrium one (same
p,
T,
V,
c, etc., for any
j), the total availability of the initial state is:
Notice that the available energy is per se not additive: each subdomain
J possesses at each instant of time 0 <
t < τ an available energy
aJ that depends on the “distance” of
J from the system final “internal” equilibrium state. The system is in internal equilibrium, though not necessarily at its dead state, at time
τ: if we introduce now the possibility that
P(
t =
τ) interacts with an external reference environment, its exergy is classically defined as the maximum work that can be extracted from this exclusive interaction:
where the sum has been maintained to signify that now the interactions may also take place at the boundary, and some internal non-uniformity (gradient of some or all of the thermodynamic quantities throughout
P) necessarily appears. Since, at time
τ, although is in
P equilibrium, the “classical” definition of exergy is valid for the entire system. Extending the above reasoning to initial non-equilibrium states, the non-equilibrium exergy of
P(
t = 0) can then be defined as:
Notice that the quantity defined by Equation (7) is always larger than the equilibrium exergy (Equation (6)), and is equal to the available energy only in the very special case in which the state of equilibrium of the isolated system is identical to the reference state (this is called the “dead state” in exergy jargon). When studying the evolution of non-equilibrium systems, Equation (7) rather than (5) or (6) should be applied.
Notice that the terms ej in (7) can also be computed directly, considering the evolution of each j-th subdomain from its initial local state δPj(t = 0) to its final dead state δPj(t = tfin) = (p0, T0, c0, V0, etc.). Notice also that when using Equation (6) or (7) a complete specification of the reference state AND of the boundary conditions must be provided.
5. Non Equilibrium Available Energy and Exergy in Solids
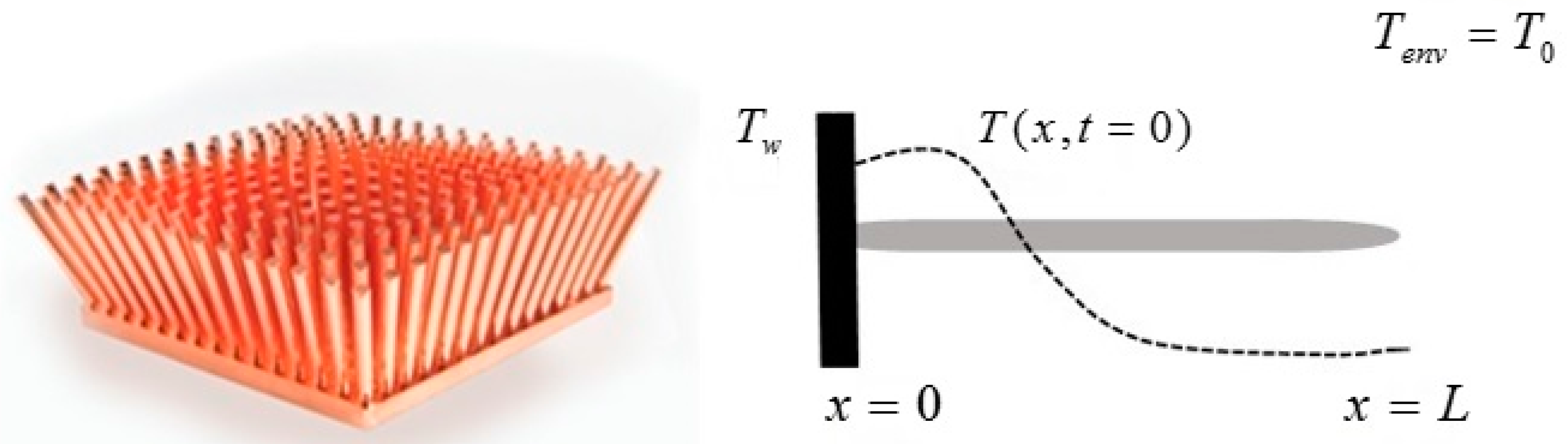
We consider a given spatially distributed mass with an initial temperature distribution and assume that the temperature evolves according to the Fourier law. The distribution of the mass can be one-, two- or three-dimensional. We shall refer in general to the mass distribution as “the solid”.
The Fourier model for the heat conduction equation in solids with convective boundary conditions reads
where
is the temperature of the environment,
is the thermal diffusivity,
is the boundary of the solid,
is the outward normal to the boundaries of the solid and
is a measure of the heat transfer by convection at the boundaries of the solid.
We remember that the available energy is the maximum work that can be extracted from a system that proceeds from an initial arbitrary state to its final internal equilibrium state (the crucial point is that the body must be isolated from its surroundings). It is well known that the maximum work associated to a heat transfer
from a temperature
to a temperature is
. In our case
is the final internal equilibrium state
(see Equation (11) below). Due to our assumption of local equilibrium,
is the local temperature at the point
at time
t of the solid. The contribution from a small element
to the availability is then given by
where
(J/K) is the heat capacity of the solid and
is the final equilibrium temperature distribution (notice that Equation (10) is, in this context, exact, since it follows directly from Gibbs’ definition, under the assumption of local equilibrium). Since, by definition, there is no exchange with the environment, we must set
in Equation (9). Then the equilibrium temperature
is found to be
where the integral must be performed over the volume (or surface, or curve)
V occupied by the solid.
Note that
can be also written, for any fixed
, as the differential of
, i.e.,
Our treatment can be generalized—with some mathematical complication—for variable density and specific heat distributions within the solid. However, in the remaining of this paper we will stipulate that
and
c, and thence
, are constant throughout the solid. If the equilibrium temperature
is taken as a reference, integrating Equation (10) from the actual temperature of the solid at
and
,
, to the reference temperature
, the contribution to the available energy is given by
The contribution from the entire solid at time
t is found by integrating over the volume (surface/curve, respectively) and taking the difference between the available energy at time 0 and the available energy at time
t:
where we assume for simplicity’s sake that both the specific heat capacity
and the material density remain constant throughout. By taking into account the conservation of energy in the insulated solid, that is
we obtain
and the total available energy is found by taking the limit
, that is
For the calculation of the exergy of the solid as a function of time, we must take the environment temperature
as reference. In addition, in the system of Equations (9), we must consider a value of
different from zero, to account for a thermal exchange between the solid and the environment. By a line of reasoning similar to the one adopted above, we derive the contribution from each particle of solid at location
and time
:
By integrating the previous expression from the actual temperature of the bar at
and
t,
, and the reference temperature
, the local contribution to the exergy is given by
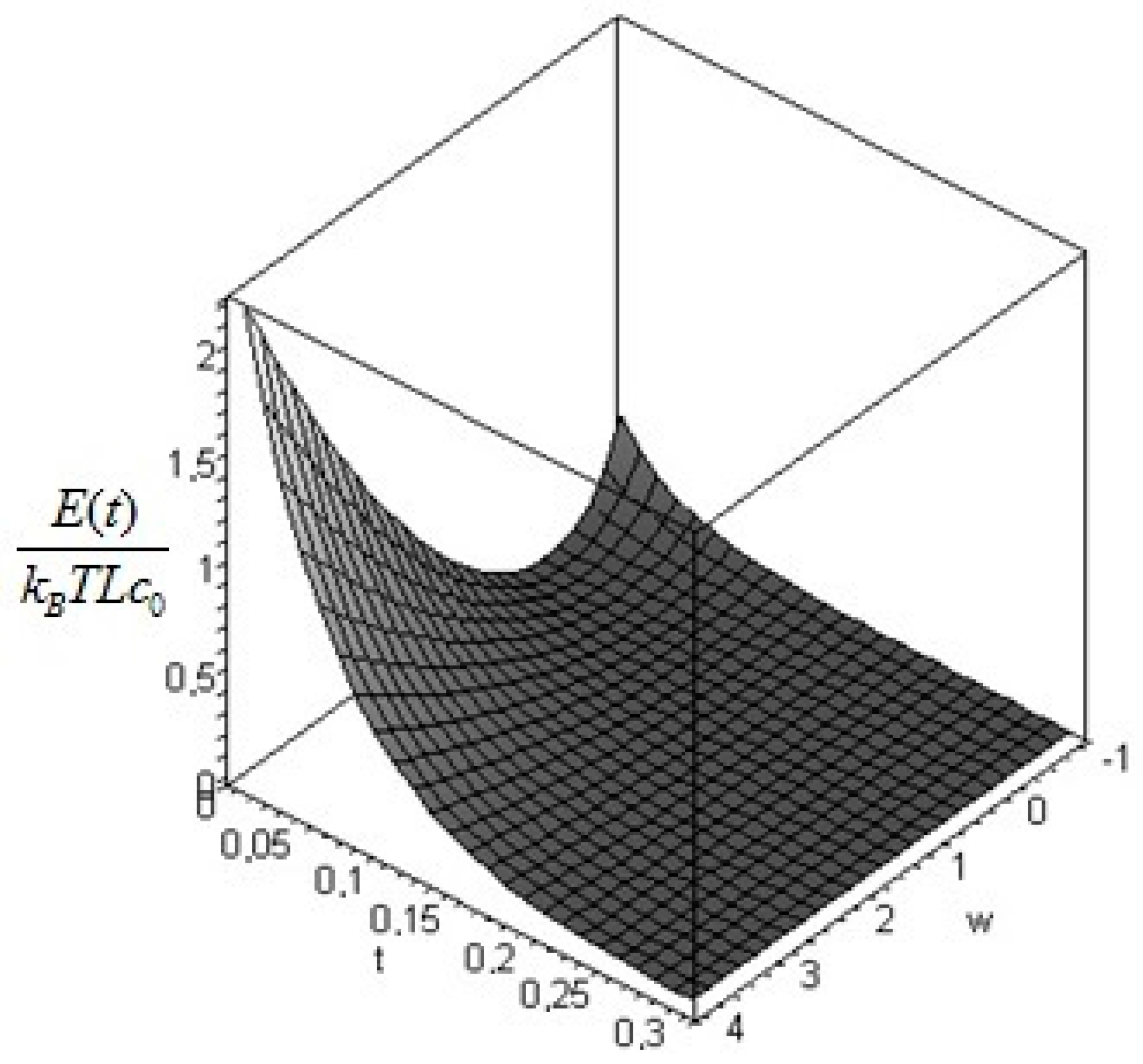
The instantaneous contribution from the entire solid at time
is found by integrating over the domain of interest, that is
where the cumulative amount of exergy destroyed at time
in the process is found by the difference between the exergy at time 0 and the exergy at time
t. We obtain:
and the total non-equilibrium exergy is found by taking the limit in
:
In this formula, is the final distribution of temperature: if the boundary conditions are described by Equation (9) (i.e., the solid is immersed in an environment at constant temperature ) then is constant and equal to .
The classical equilibrium exergy of the system is given by
Recalling that
=
, the difference between the non-equilibrium exergy
and the classical exergy
can be written, in the case of an environment at constant temperature, as:
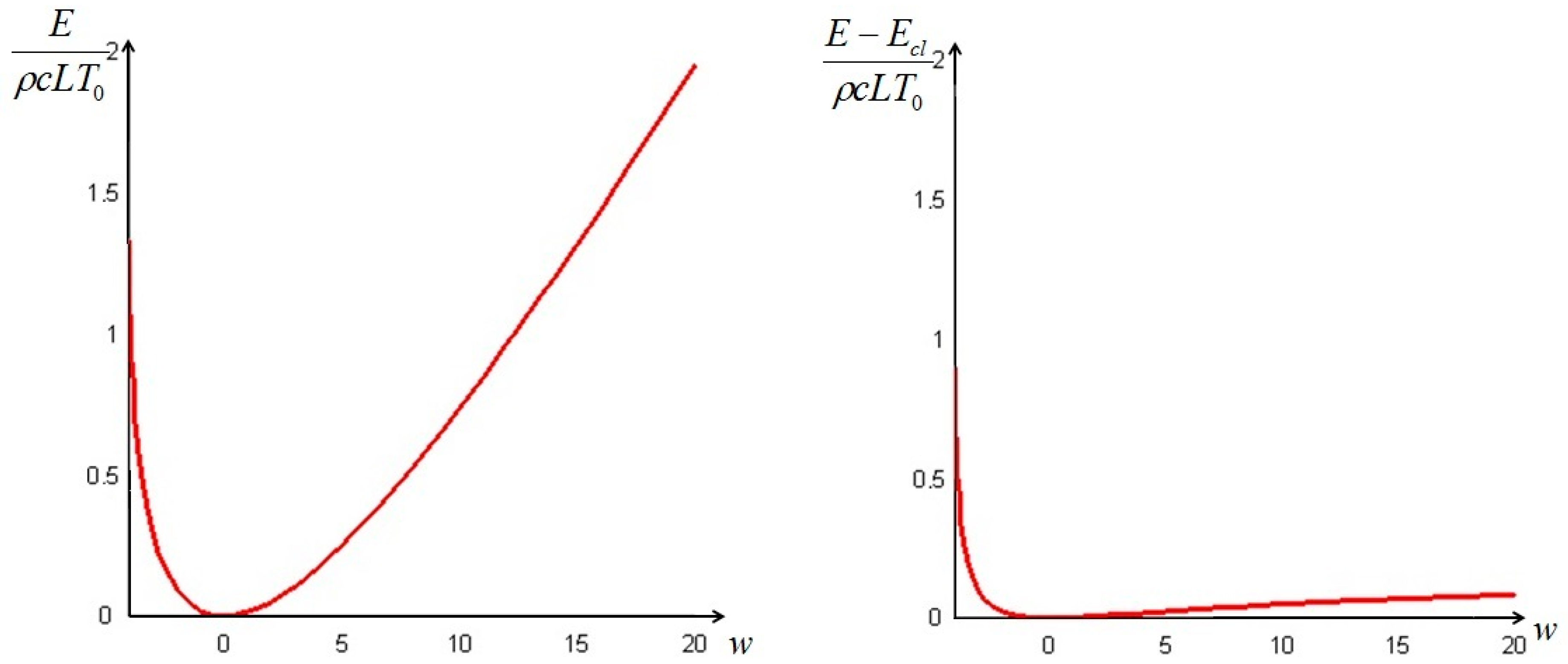
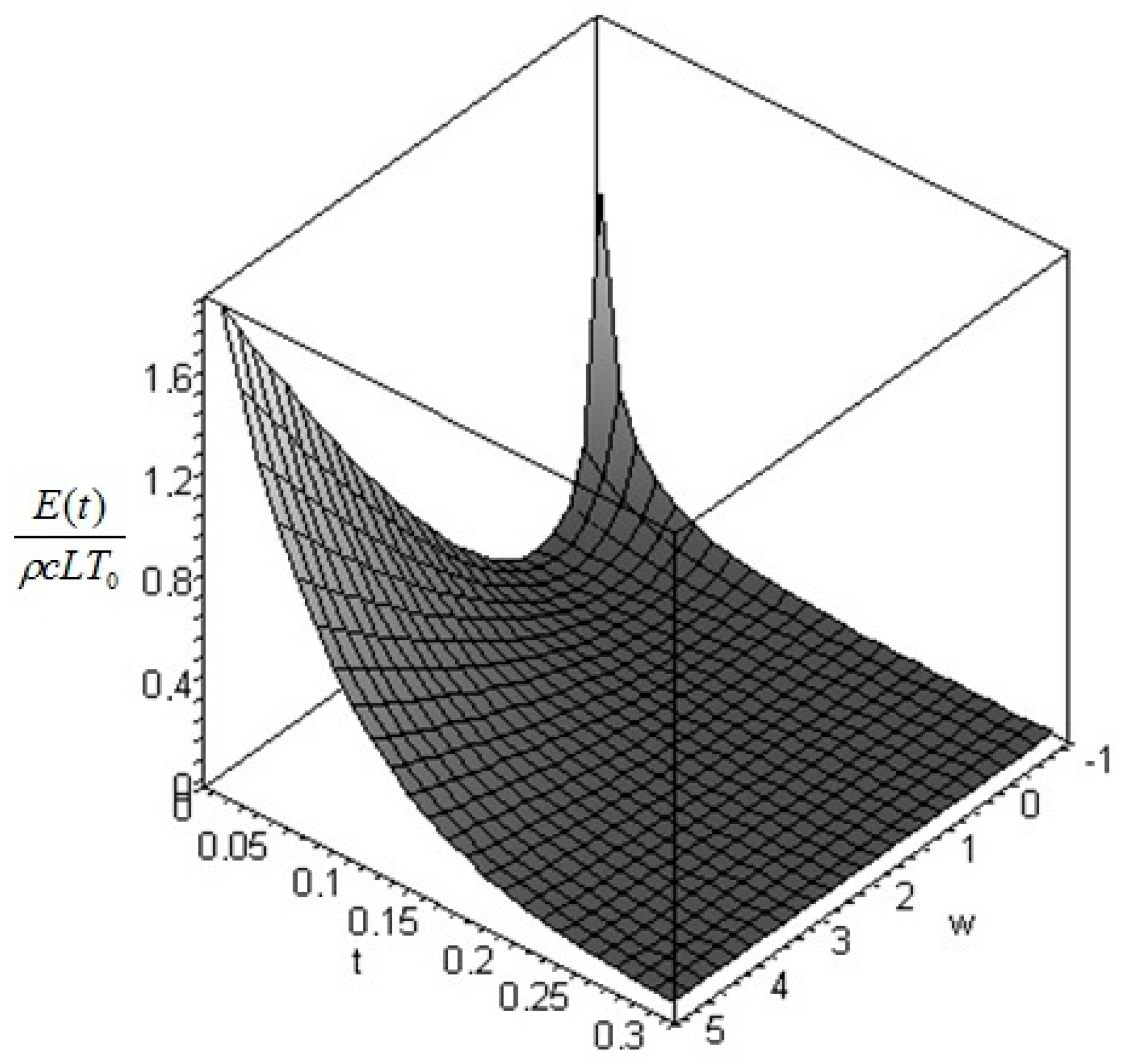
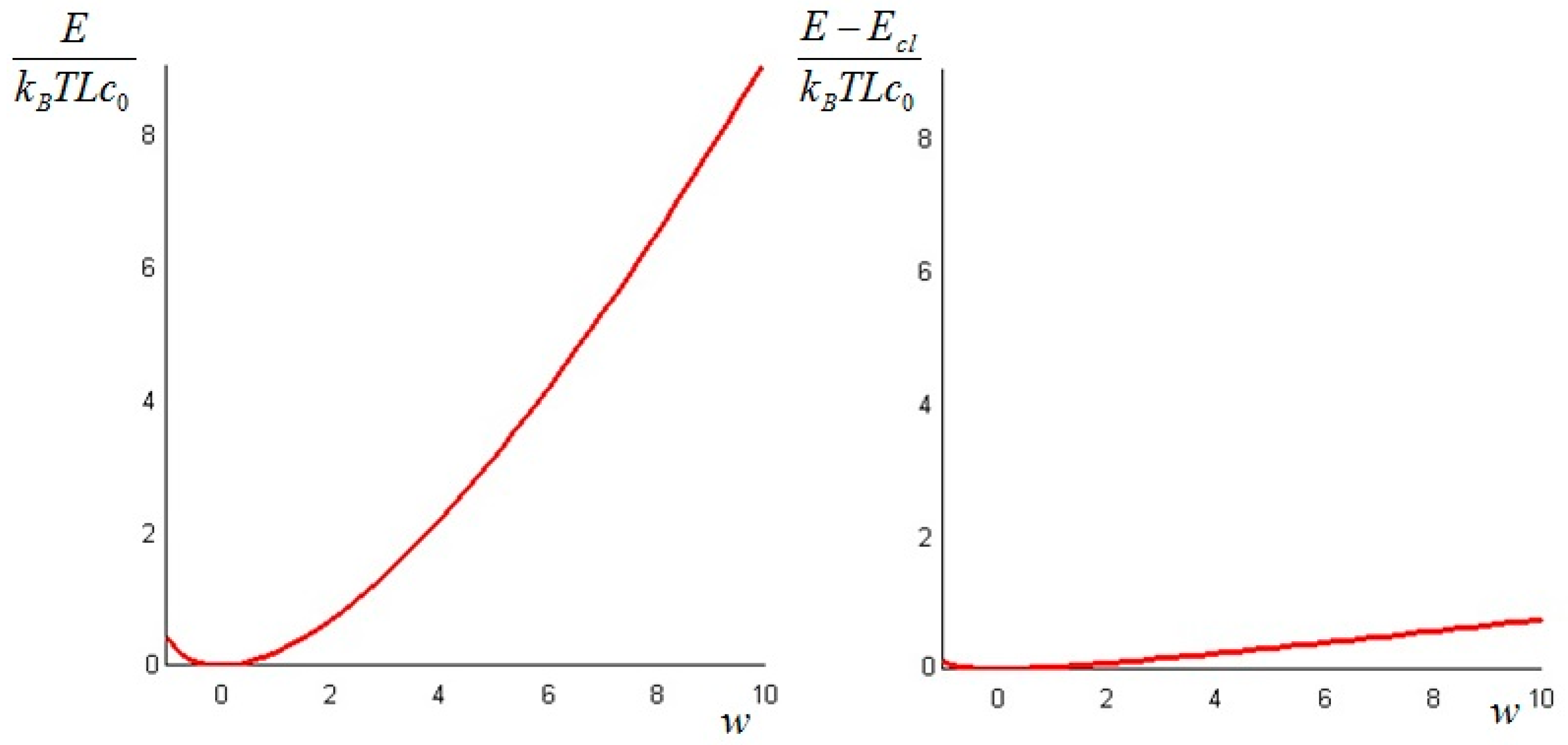
The quantity defined by Equation (24) is always nonnegative (this follows directly from Jensen’s inequality for concave functions). Thus, we can conclude that the non-equilibrium exergy (Equation (22)) is always greater than (or at most equal to) the classical exergy (Equation (23)).
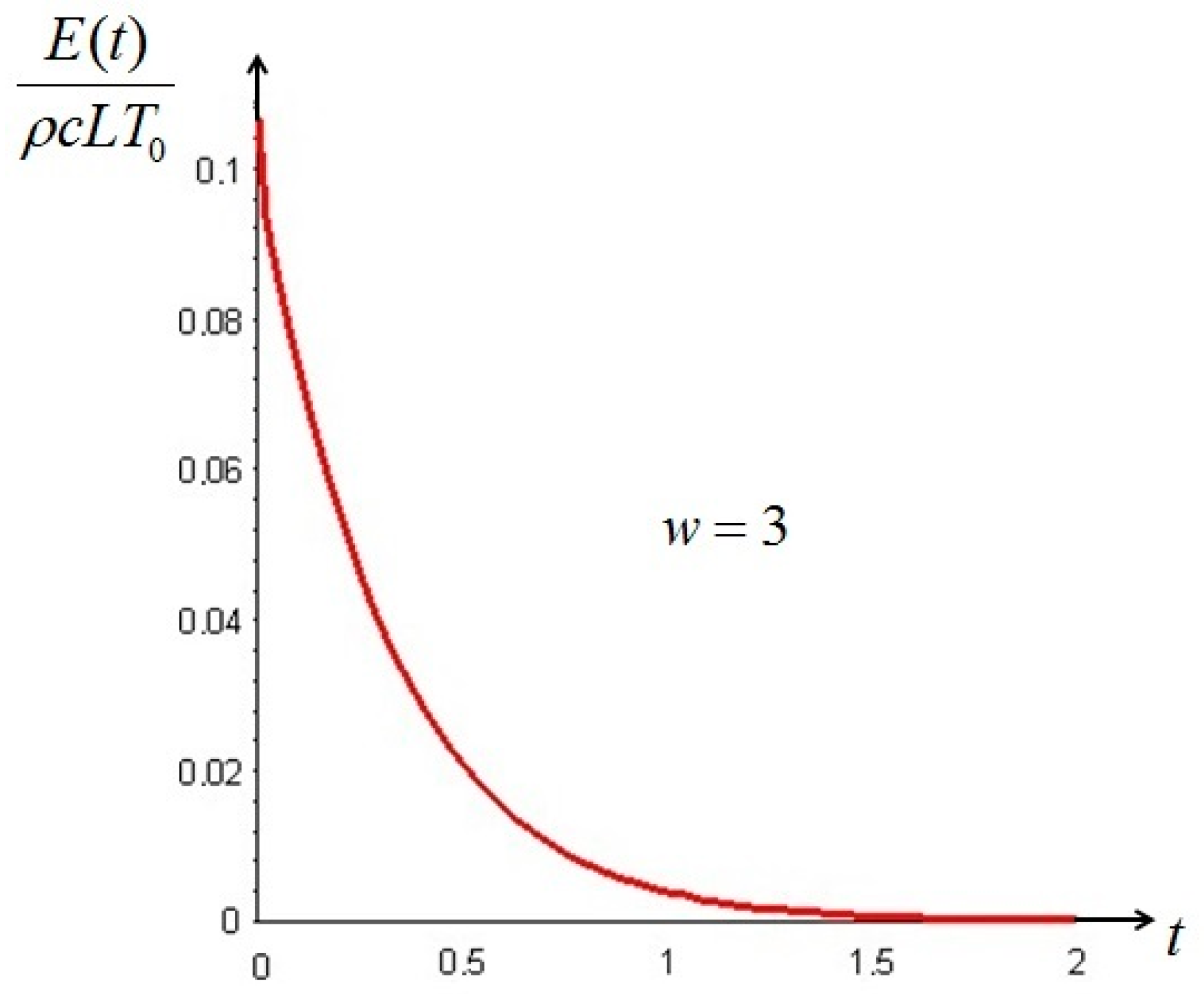
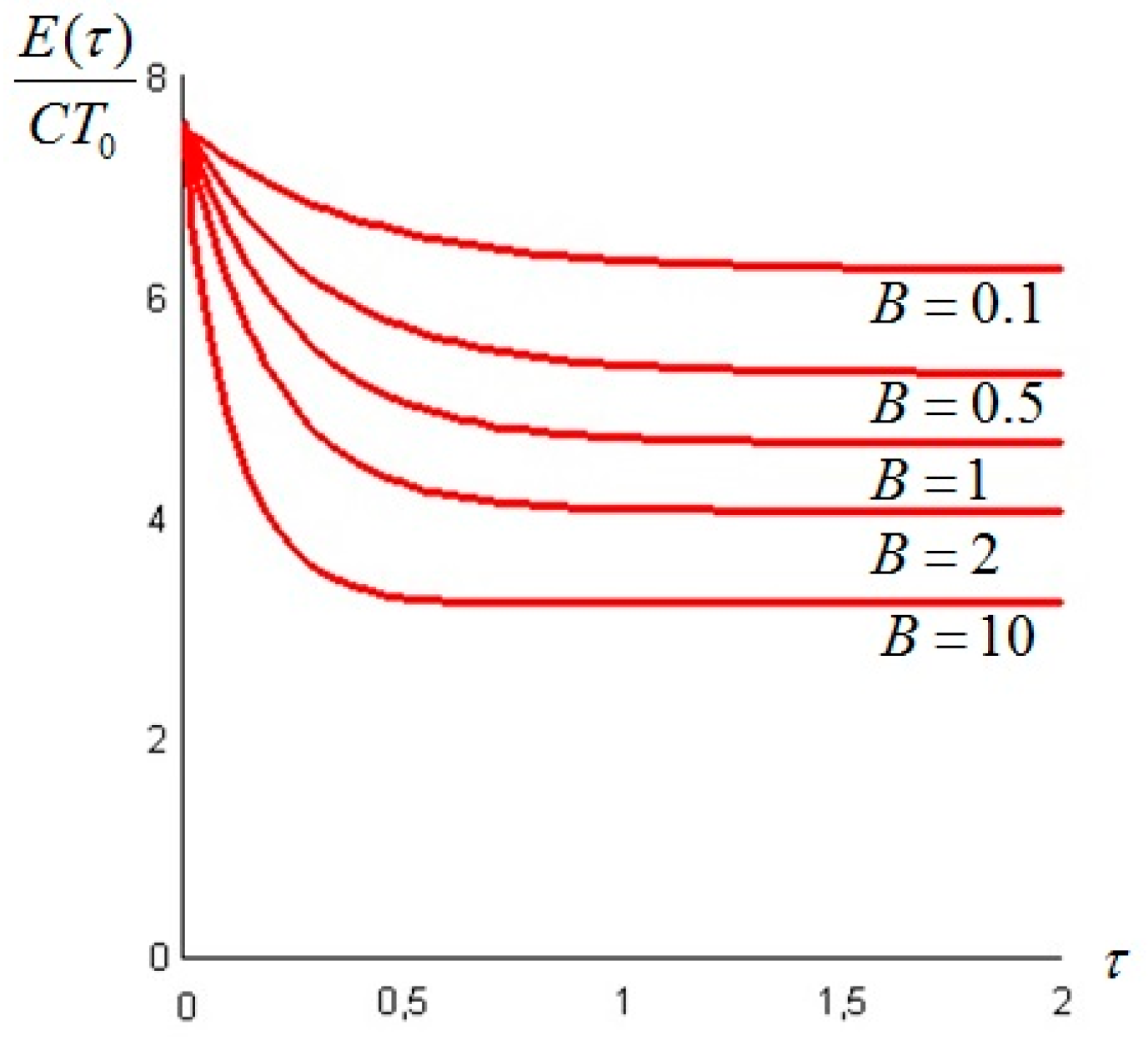
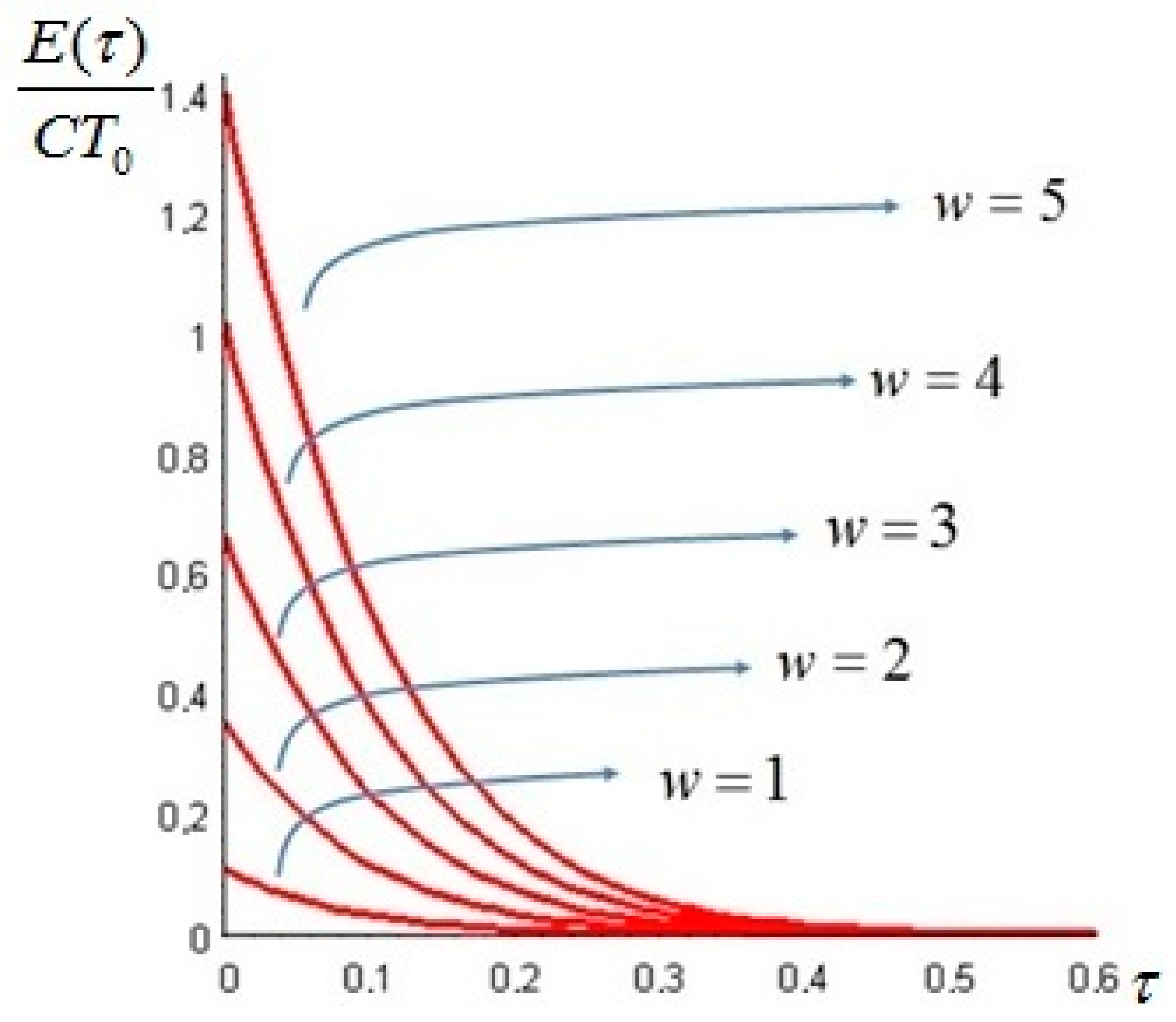
Let us make a last remark: if the steady state of the solid is a state of non-equilibrium, that is if temperature gradients on the surface of the solid still exist for
t →
∞, convective heat exchanges will also take place at large times. This can happen when the temperature on the boundaries of the solid is not uniform: for example a portion of the solid is in thermal contact with a bath at
T1 and another portion exchanges heat by convection with the environment at
T0: under these circumstances the exergy of the system will assume a constant value—different from zero—for
. An example is given in
Section 6. In such cases, it is more convenient to analyze the evolution of the irreversible exergy destruction inside of the solid: this point will be clarified in the next section.
6. A Non-Conservative Evolution Equation for Exergy
Associated with the temperature flow inside the solid there is an exergy current. This is equivalent to state that the exergy formally satisfies a non-conservative balance equation: let us discuss this point. In the absence of an internal source, the derivative of the specific exergy with respect to
, at any point in the solid, is
where we used
and
. Equation (25) follows directly from the assumptions of local equilibrium and of validity of the Fourier diffusion law. If another constitutive equation (e.g., Cattaneo’s) is taken instead,
will take a corresponding-different-form. The previous expression can be also rewritten as
If we identify the exergy flux by
(
is the heat flux), the balance equation becomes:
where
is the rate of exergy destruction per unit volume inside the solid. This term is negative definite, as it must be, because the Second Law imposes that the rate of time change in
E be always smaller than the exergy inflow through the surface of the system and always larger than the exergy outflow through that surface. From Equation (27) we see that—in a physical sense—there is no “exergy balance”: a portion of the influx is unavoidably destroyed by irreversibility. The total exergy destruction at time
is
As for the entropy balance equation, the exergy decrease of the system can be divided in two parts: one (
) is due to interactions with the environment, while the other accounts for the irreversible changes inside the system. Notice that
is related to the entropy production rate
inside the solid by
. For a discussion about the evolution and the properties of the entropy production rate due to irreversible changes inside the system see [
29,
30].
7. Non Equilibrium Exergy in Mass Transfer Processes
In this section, we consider a given initial distribution of a liquid or a gas diffusing in a region under a difference of concentrations (chemical potential). We assume that the diffusion process is described by Fick’s law.
The initial spatial distribution of concentration (number of molecules per unit of volume) in the region is a given function
and can be one-, two- or three-dimensional. We shall refer in general to this distribution as “the fluid”. The surrounding has a constant concentration equal to
and a constant temperature
T. In the case of impermeable boundary conditions, the system shall evolve towards a uniform equilibrium concentration
, given by
Let us calculate the time history of the exergy of the system. We must allow for an exchange of mass at the permeable boundaries: the contribution of the mass in
to the variation of exergy at time
is proportional to the chemical potential of the substance in
at
times the variation of the number of molecules in the region considered, i.e.,
. Indeed, since the temperature is constant throughout the body, there is no heat flow, but under the imposed b.c. there is a mass flow: the previous equation then provides, under the hypothesis of local equilibrium, a quantitative measure of the exergy content of this flow. In a completely similar way as for heat transfer processes, a corresponding value of non-equilibrium exergy may thus be derived. As the reference state for the chemical potential, we take the environmental constant concentration
. The chemical potential is given by
, where
is the activity of the substance. The concentration
is related to the activity by an activity-coefficient that we assume to be constant, so we can write [
8]
.
If
is a small region around
, the contribution in
at
is
Since we take as reference the concentration
, the contribution of the parcel
dx to the exergy of the domain is obtained by integrating Equation (30) from the concentration of the fluid at
and
to the reference concentration:
The instantaneous contribution from the entire fluid at time
is found by integrating over the volume (surface/line, respectively)
where the total amount of exergy at time
t is found by making the difference between the exergy at time 0 and the exergy at time
t:
The total non-equilibrium exergy is found by taking the limit
of Equation (33):
The classical equilibrium exergy of the system is given by
The difference between the non-equilibrium exergy
and the classical exergy
can be written as
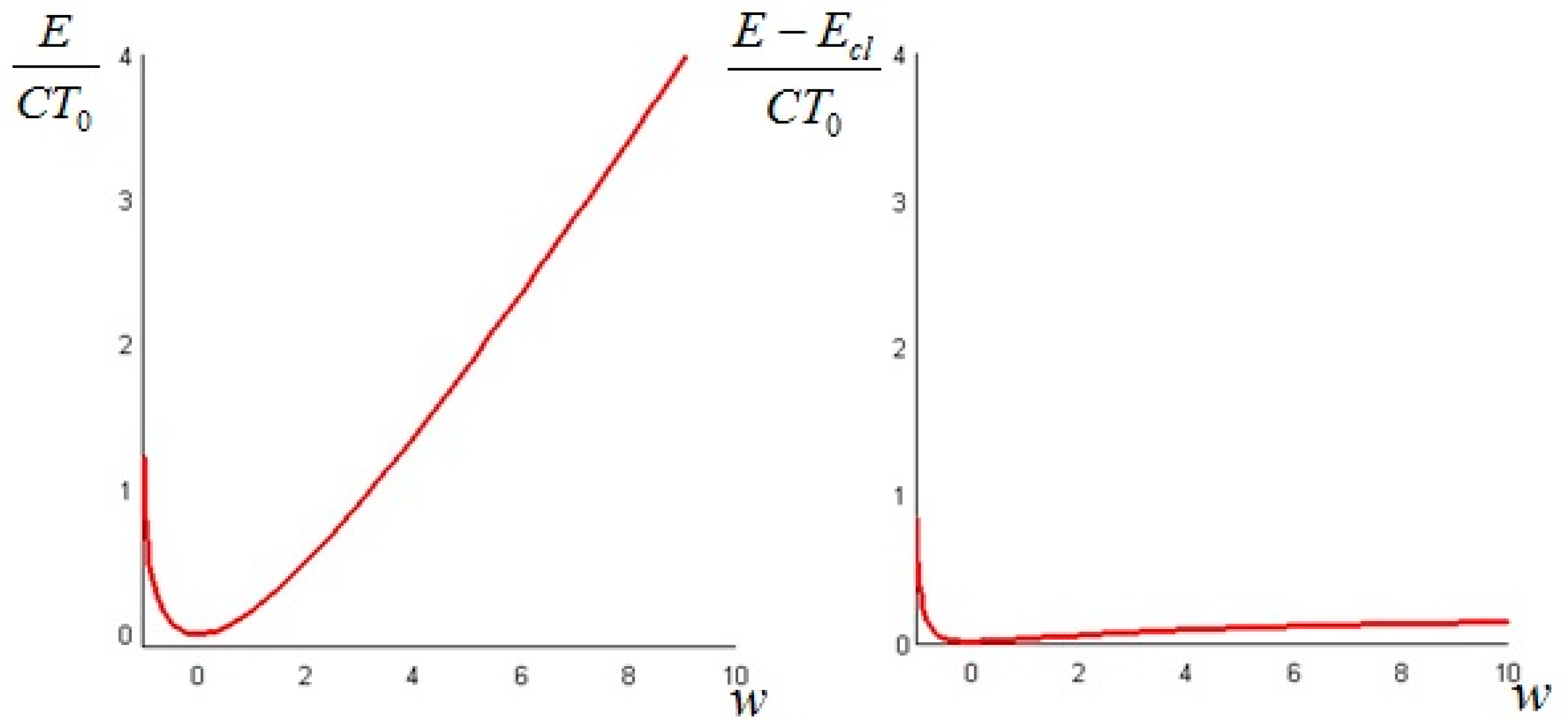
The above quantity is strictly non-negative (this follows again from Jensen’s inequality for convex functions). Hence, we conclude that the non-equilibrium exergy (Equation (34)) is always greater than (or at least equal to) the classical exergy (Equation (35)).
We can repeat the above derivation to find the expression of the available energy. In this case, we must consider impermeable boundary conditions. The available energy at time
t is be given by
where we used the conservation law
. The total available energy is found by taking the limit for
t →
∞:
In the next section, we provide examples of calculation of the non-equilibrium exergy for some physical systems of interest.
9. Conclusions
Starting from a system in thermal or concentration non-equilibrium, the evolution of the thermal or concentration fields, respectively, is obtained by analytically solving the applicable diffusion equation. Three quantities are of interest in the present context (x denotes here the proper coordinate vector):
- (1)
The Gibbs’ available energy A(t = 0), defined as the double integral (in space and time) of the quantity from x = 0 to x = X and from t = 0 to t = ∞, when the system has reached its adiabatic internal equilibrium at Teq;
- (2)
The classical equilibrium exergy E(t = 0), defined as the double integral (in space and time) of the quantity from x = 0 to x = X and from t = 0 to t = ∞; and
- (3)
The quantity we call non-equilibrium exergy En-eq(t = 0), defined as the double integral (in space and time) of the quantity from x = 0 to x = X and from t = 0 to t = ∞.
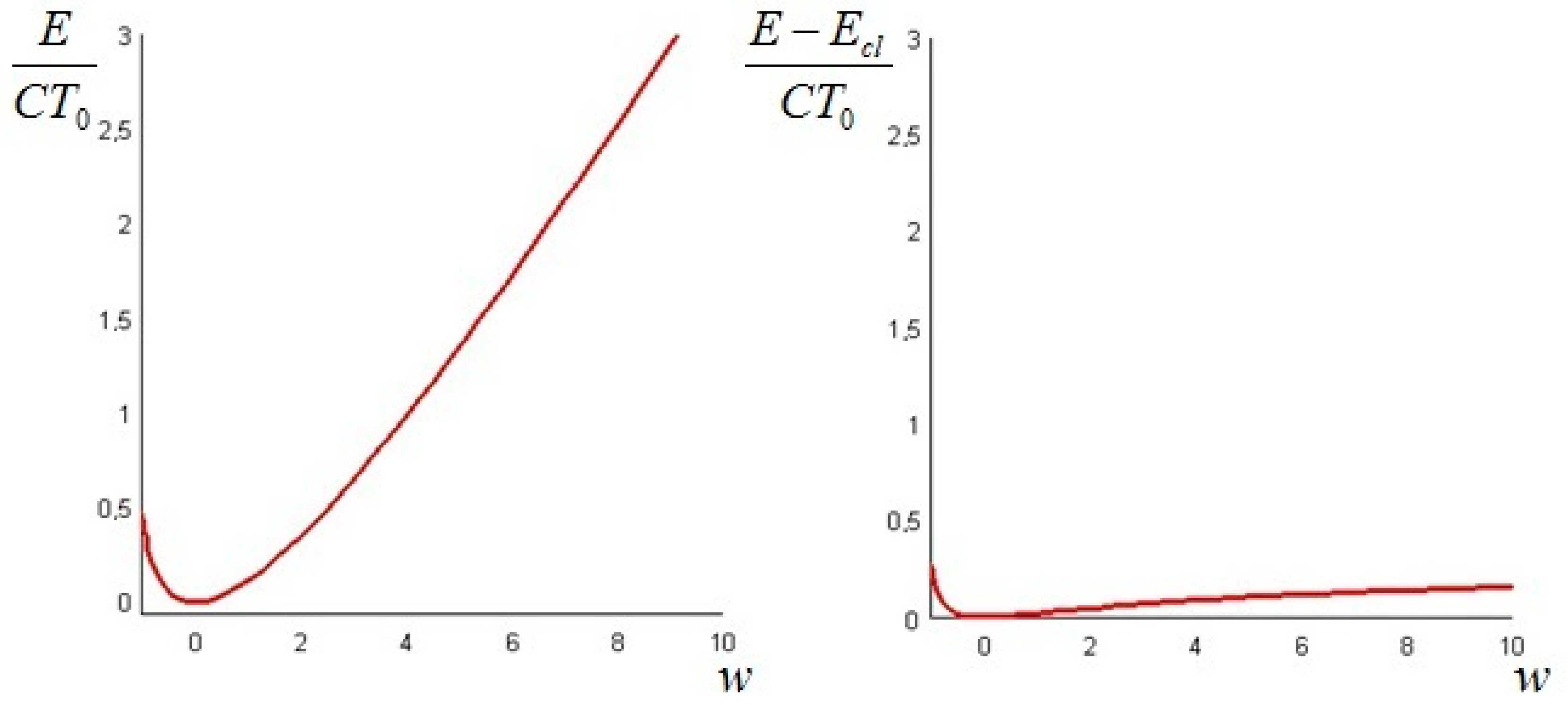
The above quantities attain different values, and while the Gibbs available energy can be directly calculated on the basis of an adiabatic energy balance (to compute the Teq), both the standard and the non-equilibrium exergy require the additional specification of a proper set of boundary conditions. The non-equilibrium exergy is always higher than its equilibrium counterpart, because it contains the portion of extra ideal work that may be extracted from the system in the course of its internal equilibration. No general relation can be derived between the two latter quantities, because the temperature (or concentration) history depend on the imposed boundary conditions and there is no a priori reason for the system to pass through its internal equilibrium state before reaching the final dead state.
Incidentally, since the initial energy level
U(
0,
x) is known and the non-equilibrium exergy
E(
t,
x) has been calculated, a non-equilibrium entropy can be defined for any system as
, its value depending on the initial
T (or
c) profile. Correspondingly, for each initial temperature or concentration distribution—and for a given set of boundary conditions—the initial entropy of the non-equilibrium system is
. The original Gibbs entropy/energy plane may be thus extended into a 3-D representation, in which each point in the “non-equilibrium” zone (that obviously corresponds in our model to a different temperature or concentration distribution) is identified by a different non-equilibrium exergy value: this topic is worthy of further investigation, and is the subject of a separate paper [
29].
As a final remark, it is important to recall that the use of the Fourier thermal diffusion law for non-equilibrium situations has been—and rightly so—criticized from a phenomenological point of view, because it implies an infinite “transport velocity” for the temperature signal. In systems with gross initial thermal inhomogeneities—however small the subdomains in which the solid is subdivided—the real non-infinite temperature diffusion velocity may bear a strong influence on the results discussed in the previous sections. If the Cattaneo transport law is used, the mathematics become slightly more involved, and at least for some of the examples discussed in this paper, analytical solutions can be found: this subject, too, will be discussed in a separate paper.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










